bigwigの作成と表示
JBrowseでbamを表示させるとリードがずらーっと並ぶ
実際にいくつあるのかわかりやすくするために
ヒストグラムで表示したい
bamをbigwig形式にする
bam2wigとwigToBigWigを使う
ここを参考にした
bamからbigWigとWiggle Formatに変換するツール - macでインフォマティクス
コマンドはここからダウンロード
bam2wig: bamからwigを作成
GitHub - MikeAxtell/bam2wig: Conversion of a BAM alignment to wiggle and bigwig coverage files, with flexible reporting options
wigToBigWig: wigからbigwigを作成
Index of /admin/exe/linux.x86_64
両方にパスを通す
wigToBigWigにパスが通っていたら
bam2wigは自動でwigToBigWigも実行する
変換はbam2wigにbamを読み込ませるだけ
$ bam2wig hogehoge.bam
こんな名前のディレクトリhogehoge_bam2wigが
作られてbigwigも出力される
できたbigwigファイルをjbrowseで表示するには
jbrowseのデータのあるディレクトリにコピーして
スクリプトで取り込ませる
How do I set up a BigWig file?
JBrowse FAQ - GMOD
$ add-bw-track.pl --label mybw --bam_url file.bw
Example BigWig-based Wiggle XY-Plot Track Configuration
JBrowse Configuration Guide - GMOD
例えば作ったファイルhogehoge.bigwigが
以下のディレクトリに存在する場合
$ /var/www/html/jbrowse/hogehoge/bw
コマンドを以下のように実行した
trackList.jsonのあるディレクトリに移動して
スクリプトを実行するのが良さそう
$ cd /var/www/html/jbrowse/hogehoge/ $ ../bin/add-bw-track.pl --label hogehoge_bigwig --bw_url bw/hogehoge.bigwig --in ./trackList.json
trackList.jsonをvimで編集
hogehoge.bigwigのあるエントリーで
typeの項目を書き換える
"type" : "JBrowse/View/Track/Wiggle/XYPlot"
登録した結果はこんな感じ

青のヒストグラムがbigwig
ヒストグラムの下の部分は
ヒトゲノムのgff3を表示している
ちなみに異なる3種類のbigwigファイルを表示しているので
ヒストグラムが3段ある
ヒトゲノムのfastaファイルとgffは
以前作ったファイルとは変わっている
前にもちょっと書いたけど公開されているヒトゲノムは
ヘッダーの書き方に違いがある
GRCh38とhg38の違い(含むミトコンドリア)
これはhisatで作ったbamファイルのラベルが変わってくる
あと、gtfファイルはUCSCのゲノムブラウザを使って作成する
Gene annotation データを用意する(gtf形式) - Palmsonntagmorgen
Table Browser
gtfファイルはgffreadを使ってgffに変換する
$ gffread -E hg38_ucsc.gtf -o- > hg38_ucsc.gff3
ちなみに逆もできる
$ gffread rename_UCSC.hg38.gff3 -T -o- > rename_UCSC.hg38.gtf
このgffファイルにはシークエンスデータのパッチも含まれる
これが意外に厄介でパッチで追加された配列に重複があると
いろいろ変換ができなくなる
問題ありそうだけど重複部分を削除することにした
良くないんだろうけど。。。
ゲノムデータのパッチの取扱がよくわからない。
重複部分の削除とJBrowseの登録はこんな感じ
$ cat UCSC.hg38.gff3 | awk '(a[$9]++ < 1000) && ($1 !~/[_alt]/) {print}' > rename_UCSC.hg38.gff3 $ bin/flatfile-to-json.pl --gff ~/usr/data/db/rename_UCSC.hg38.gff3 --trackType CanvasFeatures --out hogehoge --trackLabel ref_hg38_ch
詳しくはまたあとで。
20 April 2019追記
作り直したhg38のgtfファイルについてはここにまとめてある
mecobalamin.hatenablog.com
このファイルをJBrowseに登録したり
マッピングに使ったりした
追記ここまで
RNA-seqその6、featureCountsでリードカウント
前回作ったbamファイルを使って発現量を調べる
mecobalamin.hatenablog.com
使うコマンドはfeatureCount
こちらのサイトを参考にした
featureCount で bam と gtf からリードカウントを得る | Tips for NGS Data Analysis
https://ncrna.jp/blog/item/387-featurecount-2018
featureCountで検索かけるとRを使った解析ソフトウェア群の
Bioconductorが見つかる
Bioconductor - Home
BiocManagerというパッケージマネージャーもある。
(biocLiteはバージョンの古いマネージャー)
featureCountはRsubreadに含まれるが
こちらのwslでは動かなかった
なのでコマンドライン版のfeatureCountを使う
The Subread package
ここからダウンロードできる。
最新の1.6.3と、1.5.0-p2はこちらの環境では動かなかった
1.6.3は実行後エラーが出てすぐに止まった
1.5.0-p2はエラーメッセージが表示され続けた
1.5.3は動いたのでこのバージョンを使う
ファイルを展開後makeして$HOME/binにリンクを張った
WEHI Bioinformatics - Subread package
31 May 2019追記
最新版1.6.4が公開済みだった
アーカイブをダウンロードして展開
バイナリが含まれていたので$HOME/binにリンクを張った
ちゃんと動いた
追記ここまで
gtfファイルとbamファイルを読み込ませて実行する
結果はcounts.txtに出力される
$ featureCounts -p -O -T 4 -t exon -g gene_id -a ref_GRCh38.p12_top_level.gtf -o counts.txt hogehoge.bam
6 March 2019追記
- pと-Oオプションを追加
- pはペアエンド、-Oは重なりがあるリードがあるときに指定するらしい。
Assign reads to all their overlapping meta-features (or features if -f is specified).
featureCounts | 各遺伝子にマッピングされたリード数を計数
WEHI Bioinformatics - featureCounts
追記ここまで
このcounts.txtをRに読み込ませて解析するが、
とりあえずヒストグラムを作る
ここからはRで作業する
counts.txtのあるディレクトリに作業ディレクトリを移動し、
read.table()でファイルを読み込ませる
一行目のコメントとヘッダーを飛ばして、
タブ区切りにする
どれがカウントデータかよくわからないけど
colnames()で確認すると
[1] "Geneid" "Chr" "Start" "End" "Strand" [6] "Length" "hogehoge.bam"
なのでおそらく最後のカラム
試しにコピー数が3から30のだけを取り出して
ヒストグラムにする
setwd("/path/to/counts.txt") fc <- read.table("counts.txt", header = TRUE, skip = 1, sep="\t") d <- subset(fc, fc$hogehoge.bam < 30) d <- subset(d, d$hogehoge.bam > 3) h <- hist(d$hogehoge.bam)
結果はこんな感じ

ちなみにpng形式で保存するには
Rに表示されているグラフを消してから
png()のコマンドでファイル名とサイズを宣言
hist()を実行、そしてdev.off()でファイルを閉じる
png("histogram.png", width = 600, height = 450) h <- hist(d$hogehoge.bam, main = "", xlab= "hogehoge.bam") dev.off()
10 March 2019修正
dev.off()が抜けてたので追加
必要ならpng()の前にもdev.off()を実行して
null device
1が表示されてから上記のコマンドを実行する
hにはhistogramのパラメータが入っている
$breaksはhist()の引数breaksを指定することで
変更可能でbin幅が変えられる
> h $breaks [1] 4 6 8 10 12 14 16 18 20 22 24 26 28 30 $counts [1] 1114 358 250 196 154 134 87 104 93 64 54 51 34 $density [1] 0.206832529 0.066468622 0.046416636 0.036390642 0.028592648 0.024879317 0.016152989 [8] 0.019309320 0.017266988 0.011882659 0.010025993 0.009468994 0.006312662 $mids [1] 5 7 9 11 13 15 17 19 21 23 25 27 29 $xname [1] "d$hogehoge.bam" $equidist [1] TRUE attr(,"class") [1] "histogram"
次は目的の遺伝子がいくつあるか
グラフ化したい
11 March 2019追記
HeLaのbamファイルとhg38のgtfファイルを使ってfeatureCountsを試した
hg38のfastaとgtfはリンク先を参照
mecobalamin.hatenablog.com
HeLaのsraファイルはSRR6799791で、
hg38のシークエンスデータにhisat2を使ってマッピングした
できたファイルがHeLa.bam
featureCountsを実行する
$ featureCounts -p -B -T 4 -O -t exon -g gene_id -a rename_UCSC.hg38.gtf -o counts.txt HeLa.bam
できたcounts.txtから試しにEGFRの数を抜き出してみる
https://www.ncbi.nlm.nih.gov/gene/1956
上皮成長因子受容体 - Wikipedia
EGFRにはスプライシングバリアントがある
それぞれのaccession numberでカウントをcounts.txtから抜き出す
Rを使う
acc.egfr <- c("NM_005228", "NM_201282", "NM_201283", "NM_201284", "NM_001346897", "NM_001346898", "NM_001346899", "NM_001346900", "NM_001346941") iso.egfr <- c("a", "b", "c", "d", "e", "f", "g", "h", "i") var.egfr <- t(rbind(acc.egfr, iso.egfr)) fc <- read.table("counts.txt", header = TRUE, skip = 1, sep="\t") egfr <- c() for(i in var.egfr[,1]){egfr <- rbind(egfr, subset(fc, grepl(i, fc$Geneid)))} cbind(iso.egfr, egfr[c(1,7)])
結果はこうなったけど
これをそのままmRNAのコピー数としていいのか。。。?
iso.egfr Geneid HeLa.bam 134652 a NM_005228.4 981 134654 b NM_201282.1 254 134653 c NM_201283.1 190 134655 d NM_201284.1 255 134648 e NM_001346897.1 494 134649 f NM_001346898.1 494 134650 g NM_001346899.1 981 134657 h NM_001346900.1 963 134651 i NM_001346941.1 945
RNA-seqその5、Hisat2でマッピング
RNAseqの結果をリファレンス配列にマッピングする
mecobalamin.hatenablog.com
レファレンスデータはヒトのゲノムデータ
Human Genome Resources at NCBI - NCBI
ここからReference Genome Sequenceをダウンロード
28 Feb. 2019追記
ヒトゲノムデータにはデータの書き方の違いで2種類あるらしい
GRCh38とhg38の違い(含むミトコンドリア)
chromosomeの記述の仕方に違いがある
JBrowseで表示したり解析に使うにはhg38がいいのかも
Index of /goldenPath/hg38/bigZips
追記ここまで
20 April 2019追記
ヒトゲノムのデータを作り直した
mecobalamin.hatenablog.com
問題あるかもだけどchromosoneのファイルだけ一つにまとめて
インデックスファイルの作成に使用した
以下のスクリプトではname_dbとpostfixを書き換える
追記ここまで
まずヒトゲノムをhisat2-buildでインデックスファイルに変換する
#!/bin/bash echo "hisat2 build began" dir_scripts="$HOME/usr/scripts" dir_working="$HOME/usr/data" dir_db="${dir_working}/db" name_db="GRCh38_latest_genomic" postfix="fna" hisat2-build ${dir_db}/${name_db}.${postfix} ${name_db}
input file + 数字 + .ht2という形式で複数のファイルが出力される
GRCh38_latest_genomic.1.ht2
今回は8個だったけどいつも同じファイル数か不明
インデックスファイルができたらいよいよマッピング
hisat2の引数にインデックスファイルのパスと
ファイル名の数字の前までを指定する
マージしたFastqもリード1、2のオプションを付けて読み込ませる
samファイルで出力される
マッピングが終わるとsamtoolsでsamからbamにして
ソートしてindexをつける
stringtieでアノテーションを付ける、
とのことなんだけどよくわかってない
出力されたgtfファイルを見ると
bamに含まれる遺伝子名や場所をまとめたファイルのようだ
transcripだけ読めばカウントできそうだけどどうだろう
#!/bin/bash echo "hisat2 analysis" echo "set directories" dir_scripts="$HOME/usr/scripts" dir_working="$HOME/usr/data" dir_db="${dir_working}/db" name_db="GRCh38_latest_genomic" echo "open list sra" dir_list_sra=${dir_scripts} name_sra="list_sra.txt" list_filename=$(cat ${dir_list_sra}/${name_sra} | sed 's/\.sra//g') for i in ${list_filename} do echo "analyze ${i}" dir_out="hisat_${i}" mkdir -p ${dir_working}/${dir_out} echo "hisat began" hisat2 \ -p 4 \ -x ${dir_db}/${name_db} \ --rna-strandness RF \ --dta \ -1 ${dir_working}/trim_${i}/ps/ps_${i}_gd_1.fastq \ -2 ${dir_working}/trim_${i}/ps/ps_${i}_gd_2.fastq \ -S ${dir_working}/${dir_out}/hisat_${i}.sam \ echo "samtools began" samtools view -bS -@ 4 ${dir_working}/${dir_out}/hisat_${i}.sam |\ samtools fixmate -@ 4 - ${dir_working}/${dir_out}/fixmate_${i}.bam samtools sort ${dir_working}/${dir_out}/fixmate_${i}.bam \ -o ${dir_working}/${dir_out}/sort_${i}.bam \ -m 10G \ -@ 4 samtools index ${dir_working}/${dir_out}/sort_${i}.bam echo "stringtie began" stringtie \ ${dir_working}/${dir_out}/sort_${i}.bam \ -p 4 \ -o ${dir_working}/${dir_out}/stringtie_${i}.gtf \ -l stiringtie_${i} echo "analysis finished" done
こうやってできたbamファイルをJBrowseで表示した
18 April 2019追記
ファイルのサイズが大きい時にエラーが出てスクリプトが止まる
samtool sort の時とstringtieで起きた
samtool sortのメモリ指定を変えたほうがいいかも
ヘルプによると-m optionはmemory per threadとのこと
Windowsを起動して空きメモリは16〜20ぐらいなので
4Gとか5Gを指定するのがいいかも
試した感じでは止まらずに動いた
Stringtieは-cと-jオプションを使うといいらしい
github.com
どの程度の値がちょうど良いのか
生物学的な意味があるかもしれないが
とりあえず倍にした
-c 5 (default 2.5) -j 2 (default 1)
これでプログラム的にはオッケー
止まっていたファイルでも動くようになった
RNA-seqその4、Fastqファイルのマージ
クリーニングしたFastqファイルを一つにまとめる
mecobalamin.hatenablog.com
plinseq-liteで評価の良かったリードだけをマージする
ファイル名に"gd"を含むファイルを使うことになる
read 1とread 2を区別する
マージしたファイルは、マージ前と同じディレクトリに保存される
#!/bin/bash dir_scripts="$HOME/usr/scripts/" dir_working="$HOME/usr/data/" dir_list_sra=${dir_scripts} name_sra="list_sra.txt" list_filename=$(cat ${dir_list_sra}/${name_sra} | sed 's/\.sra//g') for i in ${list_filename} do dir_div_file="${dir_working}/raw/div_${i}" list_div_file="list_div_${i}.txt" list_read_1=$(cat ${dir_div_file}/${list_div_file} | grep _1_) list_read_2=$(cat ${dir_div_file}/${list_div_file} | grep _2_) echo "merge read 1" :> ${dir_working}/trim_${i}/ps/ps_${i}_gd_1.fastq for j in ${list_read_1} do cat ${dir_working}/trim_${i}/ps/ps_${j/_1_/_}_gd_1.fastq >> \ ${dir_working}/trim_${i}/ps/ps_${i}_gd_1.fastq #echo ${i}, ${j/_1_/_} done echo "merge read 2" :> ${dir_working}/trim_${i}/ps/ps_${i}_gd_2.fastq for j in ${list_read_2} do cat ${dir_working}/trim_${i}/ps/ps_${j/_2_/_}_gd_2.fastq >> \ ${dir_working}/trim_${i}/ps/ps_${i}_gd_2.fastq done done
14 April 2019追記
複数のファイルを処理していたら
trimmomaticのあとディスクの容量が足りなくなった
いくつか中間ファイルを消していたら
rawのディレクトリを間違って消してしまって
一緒に分割ファイルのリストも無くなった。。。
次に進めず。。。。
分割ファイルを再作成するスクリプト
mkdir $HOME/usr/data/raw/ f="$HOME/usr/scripts/list_sra.txt" fn=$(cat ${f} | sed 's/\.sra//g') for i in ${fn}; do mkdir $HOME/usr/data/raw/div_${i}; done for i in ${fn}; do for j in {1..2}; do for k in 01 02 03 04 05 06 07 08 09 10; do :> $HOME/usr/data/raw/div_${i}/div_${i}_${j}_${k}; done; done; done for i in ${fn}; do ls $HOME/usr/data/raw/div_${i}/ > $HOME/usr/data/raw/div_${i}/list_div_${i}.txt; done
空のファイル群を作成raw/に作成したあとに
ファイルのリストを作成する
あまりきれいな書き方ではないが一応大丈夫っぽい
コマンドライン上で動かしていたのでfor文も一行で書いている
このままシェルスクリプトファイルにしても動くかも
追記ここまで
20 April 2019追記
別の処理中にペアエンドのリードの数が1と2で違っている雰囲気があったので確認
前にも確認してたけど念のためもう一度
あとスクリプトの記録
$ cat hogehoge_gd_1.fastq | wc -l 61570060 $ cat hogehoge_gd_2.fastq | wc -l 61570060
リードの数は同じ
4の倍数かどうかも確認
割り算だと整数で返してくるので余りを求める
$ echo $((61570060%4)) 0
いくつかマージ後のファイルを調べたけど大丈夫そう
マージ前は?
$ for i in 01 02 03 04 05 06 07 08 09 10; do echo $(($(cat hogehoge_${i}_gd_1.fastq | wc -l)%4)); done
こっちも問題なさそう
追記ここまで
RNA-seqその3、trimmomatic
分割したFastqファイルのクリーニングをする
mecobalamin.hatenablog.com
trimmomaticについて
Trimmomatic | FASTQ クリーニングツール
trimmomaticを実行するとファイルへのアクセスがとても多い
windowsのタスクマネージャーを見ると
解析するデータが大きいのもあって
ディスクが100%近くでアクティブになる
HDDには良くない感じに見える
まず下準備としてRAMディスクを用意する
これは壊れても交換しやすいディスクがあれば別にいらないと思う
ubuntuには標準でRAMディスクがあるが、
wslのtmpfsはエミュレーションしているだけらしい
Does tmpfs really work? : bashonubuntuonwindows
なのでwindowsで動くRAMディスクをインストールする
「ImDisk Virtual Disk Driver」システムメモリ上に仮想ディスクを作成できるツール - 窓の杜
インストールしたけど場所がわからない。。。。
コントロールパネルで表示を小さいアイコンにすると
ImDisk Virtual Disk Driverのアイコンが見つかるので
ショートカットをデスクトップに作っておく
PCを再起動後にImDiskを実行する
wslを起動した状態だとマウントできないみたい
10GのRAMディスクを作成
ドライブ名はR


wslのターミナルを起動して
dfコマンドでRAMディスクにマウントできていることを確認する
マウント先はImDiskで設定したR
$ df Filesystem 1K-blocks Used Available Use% Mounted on R: 10485756 1432936 9052820 14% /mnt/r
ImDiskでボリュームを削除してもマウントが残ってたりする
RAMディスクを作ったり消したりしたらPCの再起動をしとくのが良さげ
RAMディスクを作ってマウントできてなければ手動でマウントする
それとImDiskで削除する前にアンマウントする
それぞれのコマンドはこれ
$ sudo mount -t drvfs r: /mnt/r $ sudo umount /mnt/r
作ったRAMディスクをテンポラリディスクとして使用する
ここから本題
trimmomaticの処理は以下の流れで行う
- copy files to tmp directory
- fastqc
- trimmomatic
- fastq-stats
- remove unnecessary string
- prinseq-lite
trimmomaticの前後でやっているのはシークエンスデータの統計データの取得
クリーニングの前後でデータの比較を行うための処理をやっている、
そうなんだけど見てもまだよくわからない。。。。
とりあえず使ったスクリプトだけ掲載
アダプターが取り除かれた配列が
"trim_"のついたディレクトリに保存される
#!/bin/bash cd $HOME/usr/scripts/ w_dir="$HOME/usr/data" scripts_dir="$HOME/usr/scripts" tmp_dir="/mnt/r/tmp" list_name="list_sra.txt" list_dir="${scripts_dir}" list_raw=$(cat ${list_dir}/${list_name} | wc -l) list_file=$(cat ${list_dir}/${list_name} | sed 's/\.sra//g' ) postfix="fastq" quality="15" adapter="$HOME/miniconda3/share/trimmomatic-0.38-1/adapters/TruSeq3-PE.fa" for i in ${list_file} do echo "analyze ${i}" trim_dir="${w_dir}/trim_${i}/trim" stats_dir="${w_dir}/trim_${i}/stats" fastqc_dir="${w_dir}/trim_${i}/fastqc" prinseq_dir="${w_dir}/trim_${i}/ps" mkdir -p ${trim_dir} mkdir -p ${stats_dir} mkdir -p ${fastqc_dir} mkdir -p ${prinseq_dir} mkdir -p ${tmp_dir} in_dir="${w_dir}/raw/div_${i}" div_list="${in_dir}/list_div_${i}.txt" infile_1=$(grep _1_ ${div_list}) for j in ${infile_1} do infile_2=${j/_1_/_2_} logfile=${j/_1_/_} echo "copy files to tmp directory" cp ${in_dir}/${j} ${tmp_dir}/ cp ${in_dir}/${infile_2} ${tmp_dir}/ echo "fastqc began" for k in ${j} ${infile_2} do fastqc --nogroup -o ${fastqc_dir} ${tmp_dir}/${k} done echo "finished." echo "trimmomatic began" trimmomatic \ PE \ ${tmp_dir}/${j} \ ${tmp_dir}/${infile_2} \ ${tmp_dir}/trim${quality}_${j}.${postfix} \ ${tmp_dir}/trim${quality}_${j}.${postfix}-unique.out \ ${tmp_dir}/trim${quality}_${infile_2}.${postfix} \ ${tmp_dir}/trim${quality}_${infile_2}.${postfix}-unique.out \ ILLUMINACLIP:${adapter}:2:30:10 \ LEADING:3 \ TRAILING:3 \ SLIDINGWINDOW:4:${quality} MINLEN:36 \ &> ${trim_dir}/trim_${logfile}.log echo "finished." echo "fastq-stats began" for k in ${j} ${infile_2} do fastq-stats \ ${tmp_dir}/${k} >\ ${stats_dir}/raw_${k}.stats fastq-stats \ ${tmp_dir}/trim${quality}_${k}.${postfix} >\ ${stats_dir}/trim_${k}.stats done echo "finished." rm ${tmp_dir}/${j} rm ${tmp_dir}/${infile_2} echo "remove unnecessary string" for k in ${j} ${infile_2} do #sed -i".bak" '/length/s/^\+.*/+/' ${tmp_dir}/trim${quality}_${k}.${postfix} #mv ${tmp_dir}/trim${quality}_${k}.${postfix} ${tmp_dir}/rename_${k}.${postfix} #mv ${tmp_dir}/trim${quality}_${k}.${postfix}.bak ${tmp_dir}/trim${quality}_${k}.${postfix} cat ${tmp_dir}/trim${quality}_${k}.${postfix} | sed '/length/s/^\+.*/+/' \ > ${tmp_dir}/rename_${k}.${postfix} done echo "finished." mv ${tmp_dir}/trim${quality}_* ${trim_dir}/ echo "prinseq-lite began" prinseq-lite.pl \ -fastq ${tmp_dir}/rename_${j}.${postfix} \ -fastq2 ${tmp_dir}/rename_${infile_2}.${postfix} \ -out_format 3 \ -derep 24 \ -log ${prinseq_dir}/${logfile}.log \ -out_good ${tmp_dir}/ps_${logfile}_gd \ -out_bad ${tmp_dir}/ps_${logfile}_bd \ -trim_tail_right 5 \ -min_len 30 mv ${tmp_dir}/rename_* ${trim_dir} mv ${tmp_dir}/ps_${logfile}_* ${prinseq_dir} done rm -r ${tmp_dir} done
27 Feb. 2019修正
adapterのファイル名をSEからPEに修正した
14 April 2019追記
ファイルの容量がRAMディスクよりも大きいと
エラーを吐かずにスクリプトが止まる
今回は10GBでは足りず、16GBまで増やす必要があった
RAMディスクの容量を変えたあとに
PCの再起動をせずにスクリプトを実行したところ
ディスクをうまく認識できなかったみたいで
一瞬ファイルの書き込みがあって
実行中のまま反応がなくなった
その後が変な状況になってしまった
crtl + cでスクリプトは止められず
tmuxを終了してもjobが残っていたようだ
ターミナルを終了してPCの再起動をしても
ubuntuは生きていたようで強制終了するかの選択肢がでてきた
強制終了を選んでも再起動のまましばらく先に進まず。。。
結局電源ボタン長押しで再起動
一応PCは再起動してほかも問題なく動いてくれているけれど
次回もそうだとは限らないので
RAMディスク周りをいじるときは
その前後でPCとターミナルの再起動をしておく
そもそもRAMディスクを使うのは
データを入れているHDDの保護のためなので、
テンポラリに使えるディスクがあればいい
USBメモリとか?
気になるのは書き込み速度
あとで試す
追記ここまで
30 May 2019追記
余分な文字列を削除するコマンドのところで
やり直してみたらエラーが出る
sedのテンポラリファイルを移動か削除のときにパーミッションエラーが出てる
コメントアウトして別の方法でやってみたらうまくできたみたいなのでそっちも残す
追記ここまで
JBrowseでbamファイルを表示する
インストールしたJBrowseにゲノムデータを表示させる
表示させるのは
レファレンスにするヒトのゲノムデータと
GRCh38_latest_genomic.fna
ref_GRCh38.p12_top_level.gff3
HeLaのRNAseqのデータから作ったbamファイル
こっちが実験データの代わりになる
sort_SRR6799791.bam
sort_SRR6799791.bam.bai
の四種類
bamファイルの作り方は後ほど記録する
JBrowseへの登録はこっちに説明があるので
ここからやったことを記録してく
JBrowse Configuration Guide - GMOD
JBrowse FAQ - GMOD
まずはレファレンスデータの登録
jbrowseのprepare-refseqs.plを使う
bin/prepare-refseqs.pl --fasta <fasta file> [options]
実際に入力したのはこれ
$ cd /var/www/html/jbrowse $ bin/prepare-refseqs.pl --fasta ~/usr/data/db/GRCh38_latest_genomic.fna --out HeLa_SRR6799791_dev --trackLabel GRCh38_latest_genomic --seqType dna
"--out"オプションで表示する実験データにレファレンスを出力する
"--trackLabel"はJBrowse上での表示ラベル
"--seqType"はよくわからないけどゲノムデータなのでDNAにした
outオプションで指定したディレクトリに
seqというディレクトリが作られてその内部にも
大量のディレクトリとファイルが作られてく
結構時間が掛かる
wslを使っている場合はwindows defenderからのアクセスを切っておくほうが良い
ほっとくと書き込むファイル全部をスキャンしようとする
mecobalamin.hatenablog.com
次にgff3ファイルの登録
flatfile-to-json.plを使う
bin/flatfile-to-json.pl --[gff|gbk|bed] <flat file> --tracklabel <track name> [options]
入力したコマンドはこちら
$ bin/flatfile-to-json.pl --gff ~/usr/data/db/ref_GRCh38.p12_top_level.gff3 --trackType CanvasFeatures --out HeLa_SRR6799791_dev --trackLabel ref_GRCh38
bamとbam.baiの登録もスクリプトを使う
スクリプト名はadd-bam-track.pl
baiファイルはbamと同じディレクトリに入れておけば
自動で認識してくれる
bamとbam.baiはbamという名前のディレクトリに入れて
HeLa_SRR6799791_dev以下に保存する
add-bam-track.plはHeLa_SRR6799791_devの場所で実行する
$ cd HeLa_SRR6799791_dev $ ../bin/add-bam-track.pl --label HeLa_SRR6799791 --bam_url bam/sort_SRR6799791.bam --in trackList.json
これでとりあえず作業は終了
前回volvoxのデータを表示したようにwslでapache2を起動する
mecobalamin.hatenablog.com
windows側のブラウザから以下のアドレスにアクセスする
http://localhost/jbrowse/index.html?data=HeLa_SRR6799791_dev
こんな感じで表示される
上段の黄色っぽく表示されているのがヒトゲノムデータ
下段の赤・青で表示されているのがRNAseqの結果

あまり一致していないようにみえる。。。
ヒトゲノムのコード領域と培養細胞で実際に転写されている領域は結構違うものなのか。。。?
そのあたり良くわからないけど、まぁ、とりあえず表示はできた
JBrowseのインストール、三度目の正直
12 October 2020 追記
mecobalamin.hatenablog.com
JBrowseをdockerで動くようにした
最低限必要なコマンドやライブラリをインストールした
以下の記事を参照する
mecobalamin.hatenablog.com
dockerが使えていればubuntuのコンテナに
JBrowseをインストールするのが
楽かもしれないと個人的には思った
追記ここまで
JBrowseのインストールをやりなおした。
変更点はインストール先を変えたこと、
サーバーをpythonのhttp.serverから
wslのapache2に変更したことの2点。
というのも作ったbamファイルが表示できなくて
いろいろ調べていたら公式の説明にこんなのがあった
JBrowse FAQ - GMOD
Note: servers like "SimpleHTTPServer" from Python or "http-server" from NPM are generally not full featured enough to run all JBrowse features correctly (SimpleHTTPServer does not support Range queries, and http-server interprets tabix files incorrectly). RangeHTTPServer does pass the test suite for jbrowse though, so it should work for tests https://github.com/danvk/RangeHTTPServer (but not for compressed json files).
pythonのSimpleHTTPServerではだめらしい
使っていたのはhttp.server。
サンプルデータのvolvoxは表示できたけど、
自分で作ったデータはうまく表示できないのもあったので
原因はよくわからないけどよくないのかも。。。
ということで
まずはapach2のインストールから。
Window10でLAMP (Fall Creators Update版) - Qiita
$ sudo apt install apache2
ちなみにLAMPとは、Linux、Apache、MySQL、PHP/Perl/Pythonのことだそう。
LAMP(Linux+Apache+MySQL+PHP/Perl/Python)とは - IT用語辞典 e-Words
LAMP (ソフトウェアバンドル) - Wikipedia
インストールしたapache2の起動と停止は次のコマンドで行う
$ sudo service apache2 start # 起動 $ sudo service apache2 stop # 停止
こういうやり方もあるようだけどまだ試してない
Apacheの起動 - Qiita
apache2を起動後windowsにインストールされた
web browserのアドレスバーに"localhost"と入れてエンターを押す。
apache2のindex.htmlが表示されればインストールがうまくいってる
/var/www/html/index.htmlが表示されている
次にnode.jsのインストール。
ダウンロードはここから
ダウンロード | Node.js
バイナリーファイルをダウンロードして展開
$ xz -d node-v10.16.0-linux-x64.tar.xz $ tar -xvf node-v10.16.0-linux-x64.tar
"/node-v10.15.0-linux-x64/bin/"に含まれる
node、npm、npxをパスの通っているディレクトリにリンクを貼る
コマンドは$HOME/binにリンクを貼るようにしてて
.bashrcにパスを書いてある
$HOME/binにリンクを貼るときはlnコマンドを使う
$ ln -s $HOME/local/node-v10.15.0-linux-x64/bin/node $HOME/bin
同様に"node-v10.15.0-linux-x64/lib/node_modules/yarn/bin/yarn"も
$HOME/binにリンクを貼る
あと、perlは/usr/bin/perlを使う。
miniconda3にもperlがあるが、
これを使ったら何故かコンパイルエラーが出ることがあった
どうもperlの設定ファイルのパス指定がおかしい様子。
なので.bashrcでminicondaよりも前に
/usr/bin/perlが来るようにパスを書く
export PATH="$HOME/bin:/usr/bin:$HOME/miniconda3/bin:$PATH"
これらの準備が済んでからJBrowseのインストールをする
前回よりもバージョンがちょっとだけ上がってる
任意のディレクトリにアーカイブをダウンロードして展開する
今回はホームディレクトリのtmpにwgetを使ってダウンロードする
unzipで展開後してできたディレクトリを
/var/www/html/に移動する
$ cd ~/tmp $ wget https://github.com/GMOD/jbrowse/archive/1.16.3-release.zip $ unzip 1.16.3-release.zip $ sudo mv jbrowse-1.16.3-release/ /var/www/html/jbrowse
もしjbrowse/のオーナーがrootのままなら所有者を変える
インストールでsetup.shを実行するが、sudoで実行してはいけないらしい
ちなみにwhoamiは自分のユーザー名に変える
$ sudo chown -R 'whoami' jbrowse
そしてjbrowse内のsetup.shを実行する
$ cd jbrowse
$ ./setup.sh
apacheを起動してwindowsのweb browseから
volvoxのデータにアクセスできたらインストール成功
http://localhost/jbrowse/index.html?data=sample_data%2Fjson%2Fvolvox
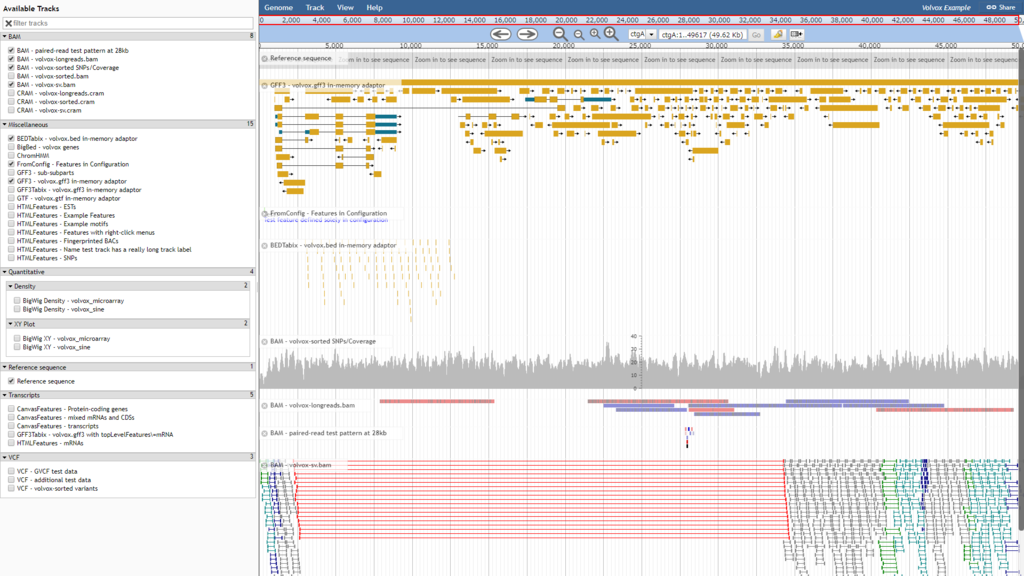
ここまで書いてなんだけど
IGVとか使えばlocal serverを立てなくても
表示できる
というかこっちが楽
サーバー立てたら共有できる点が便利
Home | Integrative Genomics Viewer
これで自作のファイルが表示できるようになればいいのだが。
25 Feb. 2019追記
apache2の起動時にエラーが表示されてた
(92)Protocol not available: AH00076: Failed to enable APR_TCP_DEFER_ACCEPT
ググると解決法があったので真似してファイルに追記した
【BoUoW】Apache起動時の「Failed to enable APR_TCP_DEFER_ACCEPT」 - Qiita
編集したファイルはapache2.conf
$ sudo vim /etc/apache2/apache2.conf
ファイルの最後に以下の二行を追加
AcceptFilter http none AcceptFilter https none
それとapache2で立てたサーバーに
ローカルネットワークからアクセスしたい
Apacheをローカルネットワークのみに公開にする | EasyRamble
サーバーになっているPCのIPアドレスと開放するポートを
ports.confに記入すればいいっぽい
$ sudo vim /etc/apache2/ports.conf
ports.confの最後の行に追記した
Listen 192.168.100.110:8080
windowsでIPアドレスを確認するには
[スタートを右クリック] -> [設定] ->
[ネットワークとインターネット] -> [Wi-Fi]
とクリックする
次に利用しているWi-FiまたはLANの名前をクリックすると
つながっているネットワークのプロパティが確認できる
IPv4アドレスをListenから:8080までの間に記入する
一応同一ネットワークにあるタブレットからvolvoxのサンプルデータを見れた
アドレスは"localhost"をファイルに追加したIPアドレスとポートに書き換える
http://192.168.100.110:8080/jbrowse/index.html?data=sample_data%2Fjson%2Fvolvox
追記ここまで
31 May 2019 追記
インストールでエラーが出る場合の対応について
ライブラリが足りなくてエラーが出る場合がある
公式のサイトに対応方法があるが、libgd2-xpm-devは見つからない
JBrowse Troubleshooting - GMOD
代わりにlibgd-devをインストールするとうまくいった
$ sudo apt-get install build-essential libpng-dev zlib1g-dev libgd-dev
追記ここまで