iOSショートカットで画像を縮小する
iPhoneで撮影した写真のサイズを変えるため
ショートカットアプリを使ってみた
切り抜きしたらサイズを変えられるけど
とりあえず画角は変えずに
元の写真から何%縮小としたい
で今回やったことは以下の2つ
相変わらずどのアクションがどんな働きをするか
この手探りがめんどい
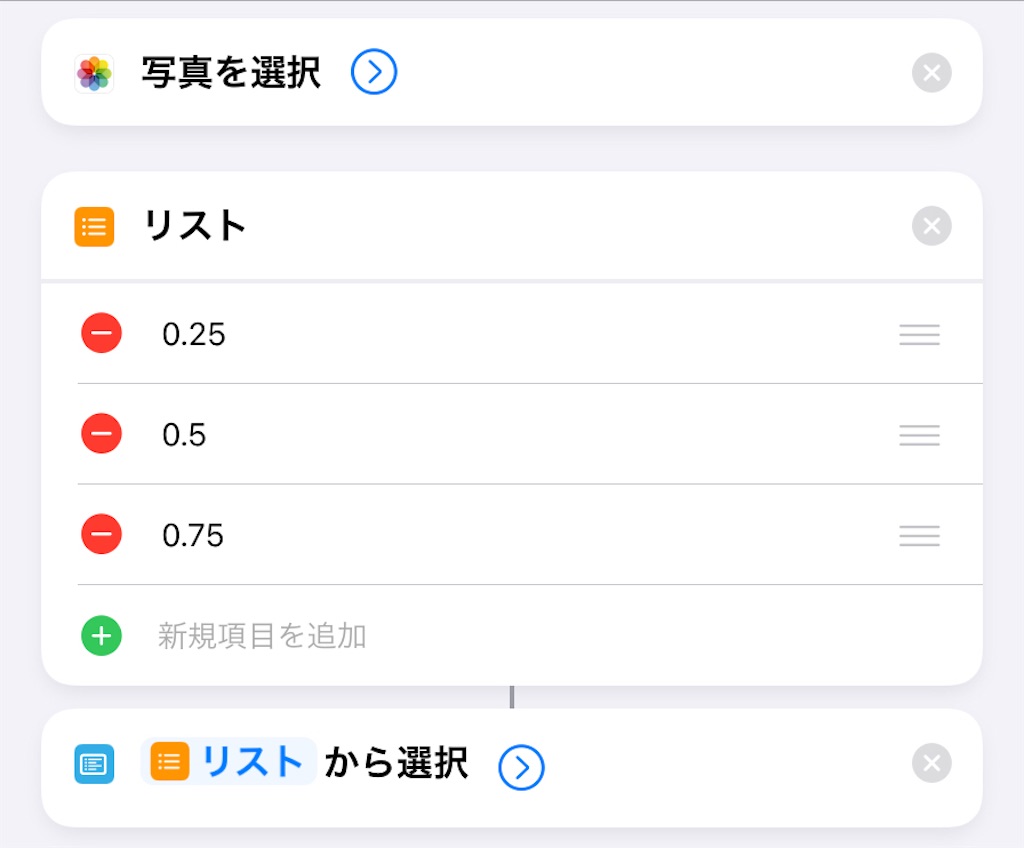
1. 保存先のアルバムを作成する
まず初めに保存先のアルバムを作成する
今回は"縮小した写真"という名前にした
iOSでアルバムとフォルダの違いがわからないが
こちらの環境だと保存先に選べるのはアルバムだった
2. ショートカットアプリでアクションを並べる
以下のようにアクションを並べる
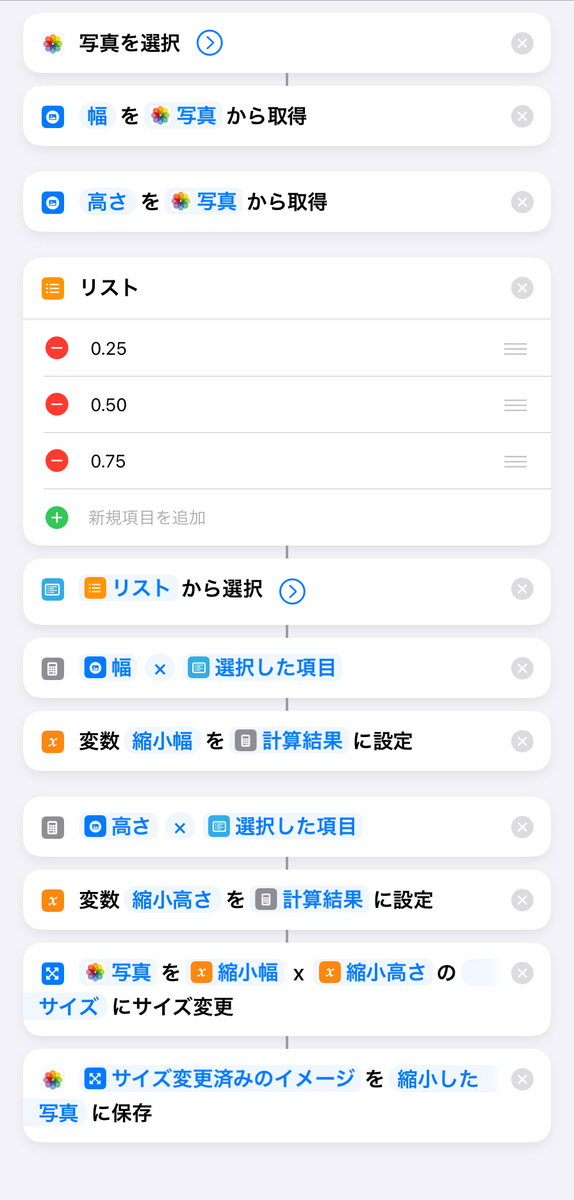
可能ならパーセント表示にしたかったが
文字列扱いのようで計算できなかった
リストで選択した文字列から%記号を除いて
数値に変換すればいいんだろうけど
取りあえず動くように簡略化した
使ったアクションは上の図を参考に検索してください
アクションの最後で画像を保存するが
1.で作成したアルバムを指定する
画像の宛先がフリーメールだとあまり気にしないけれど
受信できる添付ファイルに制限があったりするとメンドイね
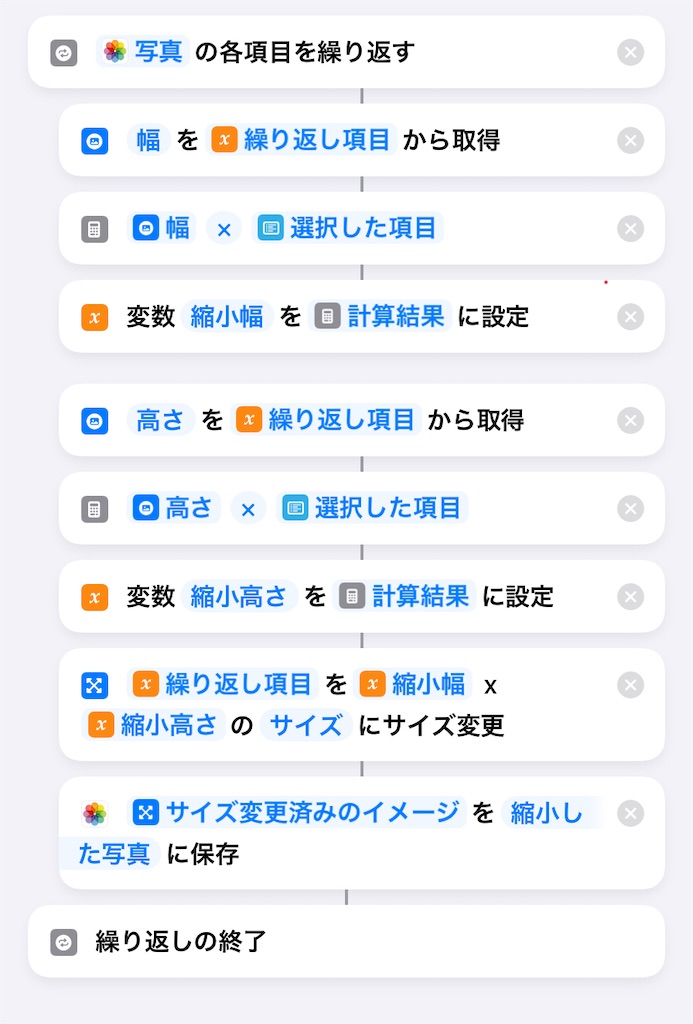
複数の写真を一括で処理するスクリプトは
以下のようになる
Vue-Routerを使ってみた
以前jQueryを使ってウェブサイトを作ってあったのだが
機能を追加するので最近のフレームワークを使ってみた
JavaScriptのフレームワークとして以下の3つが有名らしい
web開発したことないし正直どれが良いかわからない
色々調べたところこの中ではVue.js学習コストが低いらしい
そこでVue.jsでサイトを書き換えることにした
以前作ったサイトは複数のページを移動するが
実際はhtmlのファイルは一つだけで
タグのidを使って表示するコンテンツを切り替えていた
参考にしていたのはこの本
今考えるとSinge Page Application (SPA)なのかも
参考にしていた本は10年以上前に出版されていて
内容もいささか古くなっている
幸いvue.jsではVue-Routerを利用して
SPAを作ることができるそうだ
そこでVue.jsで作り直すことにした
今回は試しにVue-Routerを使ってページを表示する
やったこと
1. サーバーを立ち上げる
Vue-Routerのファイルをテストするときはサーバー経由で実行する
サーバーを経由せずにファイルを実行すると
CORS policyで実行がブロックされる
私は2種類の方法でサーバーを使っている
1-a. wslでnginxを実行する
1-b. vs codeの拡張機能でLive Serverを使う
1-a. wslでnginxを実行する
以前nginxをDocker上に起動する記事を書いた
JBrowseをDockerで動かす、その5、Nginxの起動とJBrowseへのアクセス - mecobalamin’s diary
メモ: DockerでNginxを使う - mecobalamin’s diary
これを利用している
1-b. VScodeの拡張機能でLive Serverを使う
VScodeの拡張機能にはLive Serverがあり
編集したファイルをブラウザで実行できる
Live Server実行前に作成したファイルのあるフォルダを
ワークスペースに追加する
こっちもお手軽で便利
2. .html、.vueファイルを作成する
作成するファイルは1つのhtmlファイルと(index.html)
2つのVueファイル(home.vue、about.vue)、
ひとつのJavaScriptファイル(index.js)の4つ
index.htmlファイルでCDNを読み込む
vue 3
router-vue 4
http-vue-loader
JavaScriptファイルも読み込む
Vue-Routerを使うときVue-cliを使うという説明が多いが
使い方がちょっとめんどい
http-vue-loaderを使う手もある
qiita.com
私はhttp-vue-loaderを使っている
bodyタグにidが"app"のdivタグとrouter-viewタグを記述する
Router-Vueはidが"app"のdivタグの子にあるrouter-viewタグに
.vueファイルを差し込む
index.html
<!DOCTYPE html> <html> <head> <meta charset="UTF-8" /> <title> Vue-Routerを使ってみた </title> </head> <body> <div id="app"> <div> <router-view></router-view> </div> </div> <script src="https://cdn.jsdelivr.net/npm/vue@3"></script> <script src="https://unpkg.com/vue-router@4"></script> <script src="https://unpkg.com/http-vue-loader"></script> <script src="./js/index.js"></script> </body> </html>
home.vueとabout.vueにはそれぞれtemplateタグがある
このタグの子がindex.htmlのrouter-viewタグに表示される
router-link to="/about"はブラウザで表示するとき
a href="#/about"に置き換わる
home.vue
<template> <div> <div> <h2> ここはホームです </h2> </div> <div> <router-link to="/about">このサイトの説明ページに移動します</router-link> </div> </div> </template>
about.vue
<template> <div> <div> <h2> ここはVue-Routerのテストサイトです </h2> </div> <div> <router-link to="/">ホームに戻ります</router-link> </div> </div> </template>
3. JavaScriptファイルを作成する
JavaScriptファイルにルートを定義する
pathとcomponentの組み合わせで
「pathにアクセスしたらcomponentを呼び出せ」
というふうに表す
今回の場合だと"/"で"./home.vue"を呼び出せとなる
componentはhttp-vue-loader経由で目標のファイルを呼び出す
const routes = [ { path: '/', name: 'home', component: httpVueLoader('./home.vue') }, { path: '/about', name: 'about', component: httpVueLoader('./about.vue') } ] const router = VueRouter.createRouter({ history: VueRouter.createWebHashHistory(), routes }); const app = { }; Vue.createApp(app).use(router).mount("#app");
VueRouter.createRouterでルーターを生成する
createRouterにはいくつかオプションがあるが
試した感じだとhistoryとroutesオプションは必須
historyオプションはhistory/hashモードのいずれで動作するかを指定する
違いをわかっていないがhashモードを使っている
qiita.com
routesオプションでルートを定義した変数routesを指定している
実際には「routes: routes」と書くが省略構文でroutesとだけ書いてある
通常はappにmethodsやcomputedを記述するが
今回は使用しないので空にした
最後にappをindex.htmlの#appにマウントしつつ
routerを有効化した
ファイルを作成できたらサーバー経由で実行する
とりあえずここまでできたら
複数のページを切り替えながら表示できるはず
メモ: カッコに囲まれた文字を正規表現で置換する
全角・半角のカッコに囲まれた文字列を正規表現で置換する
元の文字列はこんな感じ
text = '---(abc)(123)---';
abcを囲むのは半角カッコ
123を囲むのは全角カッコ
javascriptのreplace()を使うとき
text.replace(/[((].*[))]/g, '');
の出力は
------
カッコの中身を取り除くときはこんな感じで
text.replace(/(?<=[((]).*?(?=[))])/g, '');
出力は
---()()---
リンクの範囲を広げる
htmlでリストで並べたリンクを枠で囲むことがある

通常はカーソルでリンク先に飛ぶのは赤枠で囲った範囲
リンク範囲をcssで黄色に表示させるとこんな感じ

これを緑の枠で囲った範囲全体に広げたい

カーソルを重ねるとこんな感じ

しばらく悩んでいたが解決したのでその忘備録
やったこと
解説できるほどの知識もないので
できたソースを載せる
そのままコピペで動くはず
一行分だけリンク範囲を広げるにはdisplay: blockを指定するのだが
上下に範囲を広げるにはスタイルシートでdisplay: frexにして
親のタグ(li)と同じ大きさを子のタグ(li a)に指定する必要があった
<!DOCTYPE html> <html lang="ja"> <head> <meta charset="UTF-8"> <title>リンクの範囲を広げる</title> <style> body { font-size: 24px; } li { display: flex; border: 3px solid green; height: 4em; width: 300px; } li a { display: flex; border: 1px solid red; height: 100%; width: 100%; justify-content: center; align-items: center; } li a:hover { background-color: yellow; } </style> </head> <main> <body> <ul> <li> <a href="https://www.google.com" target="_blank">Google</a> </li> </ul> </body> </main> </html>
pythonでpdfの表を読み込む
pythonにtabula-pyというpdfから表を読み込むパッケージがある
インストールと使用時にちょっと戸惑ったのでメモ
やったこと
1. tabula-pyのインストール
pipでインストールしたが、ちょっと手間取った
というのもインストールしたいのは"tabula-py"だが
"tabula"というパッケージもあり
うっかりこちらをインストールしてしまった
"tabula"がなんのパッケージなのか調べなかったが
"tabula-py"と"tabula"では使えるコマンドが違うし
両方入れていても"tabula-py"は正常に動かなくてしばらく悩んだ
それでまず"tabula-py"と"tabula"の両方をアンインストールして
"tabula-py"を再インストールすると正常に動いた
インストールされているパッケージを確認すると
"tabula"と"tabula-py"がインストールされているので
まずは両方アンインストールした
> pip list tabula 1.0.5 tabula-py 2.6.0 > pip uninstall tabula > pip uninstall tabula-py
pipのアップグレードを行って
"tabula-py"を再インストール
> pip install --upgrade pip > pip install tabula-py > pip list sympy 1.5.1 tabula-py 2.6.0 terminado 0.8.3
とりあえずこれで準備完了
2. tabula.read()を使う
pdfの読み込みにtabula.read_pdf()を使った
引数はファイルのパスと、表の有無、読み込むページを記述した
filename.pdfから表ありの1ページ目を読み込むときには以下のように記述する
import tabula tabula.read_pdf(/path/to/filename.pdf, lattice = True, pages = 1)
importに"tabula"と記述するのがややこしい
戻り値は基本DataFrameだが
読み込んだpdfに複数の表があると
リストにまとめられている
例えば2つ目の表の1行目を取り出すには以下のようにする
tabula.read_pdf(/path/to/filename.pdf, lattice = True, pages = 1)[1].iloc[0, :]
3. CSV形式で保存
取り出した表の保存にto_csv()を使用した
df = tabula.read_pdf(/path/to/filename.pdf, lattice = True, pages = 1)[1].iloc[0, :] df.to_csv(/path/to/filename.csv, encoding = 'cp932')
windowsを使っているのでencodingにcp932を指定した
wslでgithubを使う
wslでgit hubを使えるようにした
やったこと
主にこちらのサイトを参考にさせてもらった
qiita.com
以下のGitHubのアカウントを持っているものとする
アカウント名: hogefuga
メールアドレス: hogehoge@fugafuga.com
1. gitのアップデート
新しいパッケージリストを取得後にgitをインストールする
$ sudo apt-get update $ sudo apt-get install git
2. git configの設定
こちらのサイトを参考にさせてもらった
qiita.com
wslのコマンドラインで以下のコマンドを実行する
$ git config --global user.name "hogefuga" $ git config --global user.email "hogehoge@fugafuga.com" $ git config --global pull.rebase false $ git config --global core.editor "code --wait"
1行目と2行目でgithubのアカウントとメールアドレスの登録を行う
3行目はpullの挙動に関するものらしい
4行目はvscodeをエディターとして登録する
3. sshの設定
2.で参考にしたサイトによると
SSH認証 ... GitHubに公開鍵を登録し、自分のPCに登録した秘密鍵と照合して認証する方法。初期設定さえすれば、毎回のアカウント名・トークンの入力は必要ない。
とのことなのでssh認証の設定を行った
手順としては以下の通り
3-1. sshキーを作成
3-2. sshキーをクリップボードにコピー
3-3. sshキーをgithubに登録
3-4. ssh接続を確認
3-1. sshキーを作成
wslのコマンドラインで以下のコマンドを入力する
$ ssh-keygen -t rsa
3回質問があって入力待ちになるが、デフォルトで良かったので何も入力せずにエンターキーを押した
3-2. sshキーをクリップボードにコピー
以下のコマンドでコピーする
$ cat ~/.ssh/id_rsa.pub | clip.exe
3-3. sshキーをgithubに登録
webブラウザからgithubにログインし、
[右上のアイコン] ->[settings] -> [SSH and GPG keys]の順でクリック
[SSH keys]の[new SSH key]からwslで作成したssh keyを登録する
[new SSH key]をクリックするとtitleとkeyの入力欄が現れる
titleはわかりやすい任意の名前を入力する
私は"wsl_ubuntu"とした
keyにはクリップボードにコピーしたssh keyを貼り付けた
最後に[add SSH key]のボタンを押して登録終了
3-4. ssh接続を確認
wslのコマンドラインから接続テストをする
$ ssh -T git@github.com
初回のみ警告が出て接続してよいか確認される
"yes"とタイプしてエンターでよい
最後に以下のメッセージが出たら接続できている
Hi hogefuga! You've successfully authenticated, but GitHub does not provide shell access.
4. githubにpush
実際にファイルをgithubにpushする
手順は
4-1. githubにリポジトリを作る
4-2. pushするファイルのあるディレクトリをinitする
4-3. pushするファイルをaddする
4-4. コミットメッセージの作成
4-5. ファイルをpushする
4-1. githubにリポジトリを作る
githubにログインし、右上のアイコンから[your repositories]をクリックすると作成したリポジトリの一覧が現れる
まだ何もなければリポジトリは表示されない
[new]のボタンを押すと新しくリポジトリを作成できる
リポジトリに名前をつけて"public"か"private"かを選んで新しく作成する
他の項目は今のところデフォルトのままにしてある
今回は"test"という名前で"private"のリポジトリを作成した
4-2. pushするファイルのあるディレクトリをinitする
目的のファイルのあるディレクトリに移動し、Gitのリポジトリを作成する
例えばpushしたいファイルのあるディレクトリが"/hoge/fuga/"なら
$ cd /hoge/fuga/
$ git init
とすると"/hoge/fuga/"に.gitのディレクトリが作成される
4-3. pushするファイルをaddする
次にpushするファイルをaddするのだが個別にファイルをpushする場合は
$ git add [ファイル名]
とし、ディレクトリに含まれるファイルをすべてpushする場合は
$ git add .
とする
4-4. コミットメッセージの作成
コミットメッセージとはコミット時に入力を求められる文章のことで、ファイルに対してどのような操作をしたかを簡潔に説明した文章である
$ git commit
を実行すると、2. git configで設定したようにvscodeでCOMMIT_EDITMSGが起動する
ファイルを編集して閉じるまでwslはwait状態になる
コミットメッセージには書き方があるそうで以下のサイトを参考にした
qiita.com
ファイルを閉じると次のメッセージが表示された
Aborting commit due to empty commit message.
git configでwaitオプションを付けると解決するそうだが、私の環境ではうまくいかない
k-side.hatenablog.jp
commit文は作成されているのでこれからあとの操作に問題はない
4-5. ファイルをpushする
実際にファイルを作成したリポジトリにpushする
リポジトリのアドレスは作成したリポジトリの[<> code] のタブで[<> code]のボタンを押すと[ssh]のタブが見つかる
[ssh]のタブに"git@github.com"で始まるアドレスがあるのでこれを使う
今回はユーザー名hogefugaで作成したリポジトリ名がtestなので"git@github.com:hogefuga/test.git"となる
実際にpushするまでのコマンドは以下の通り
$ git remote set-url origin git@github.com:hogefuga/test.git $ git branch -M main $ git push -u origin main
pushが終わったらウェブブラウザでファイルを確認する
5. sshプロトコル通信に変更
実際に使ってみてわかったことなのだがgit pushコマンドを使うとなぜか毎回ユーザー名とパスワードを聞かれてた
入力してもログインできずに困っていたら同じ問題を解決している人がいた
qiita.com
こちらのサイトを参考にhttps通信からsshプロトコルに変更した
まず現状の確認
$ git remote -v origin https://github.com/hogefuga/test.git (fetch) origin https://github.com/hogefuga/test.git (push)
test.gitはgit hubにあるリポジトリ
httpsを使っていたのでsshに変更する
$ git remote set-url origin git@github.com:hogefuga/test.git $ git remote -v origin git@github.com:hogefuga/test.git (fetch) origin git@github.com:hogefuga/test.git (push)
上でも書いたが、リポジトリのアドレスは目的のリポジトリの[<> code] のタブで[<> code]のボタンを押すと[ssh]のタブが見つかる
[ssh]のタブに"git@github.com"で始まるアドレスがあるのでこれを使う
[おまけ] ファイルのダウンロード
githubにあるファイルのクローンをダウンロードするにはファイルを保存したいディレクトリに移動したあとに
$ git clone https://github.com/hogefuga/test.git
または
$ git clone git@github.com:hogefuga/test.git
とする
Vue2からVue3に書き換え
Webデザインの勉強のためにVue.jsの本を読み始めたのだが初っ端でつまずいた
本に書かれているとおりにコードを書いて表示させると
{{ message }}
デベロッパーツールでエラーの場所を確認すると以下のエラーメッセージ
Uncaught TypeError: Vue is not a constructor
同じエラーで記事を書いている人がいた
minamino-heya.com
qiita.com
Vue.jpのバージョン違いで書き方が大きく変わっているらしい
読み始めた本はVue2系で書かれているようだ
先程の記事を参考にVue2とVue3で書いてみた
Vue2の場合
<!DOCTYPE html> <html lang="ja"> <head> <meta charset="UTF-8"> <title>Hello, world!</title> <script src="https://cdn.jsdelivr.net/npm/vue@2"></script> </head> <body> <div id="app"> {{ message }} </div> <script> new Vue({ el: "#app", data: { message: "Hello World!" } }); </script> </body> </html>
Vue3の場合
<!DOCTYPE html> <html lang="ja"> <head> <meta charset="UTF-8"> <title>Hello, world!</title> <script src="https://cdn.jsdelivr.net/npm/vue@3"></script> </head> <body> <div id="app"> {{ message }} </div> <script> const app = { data(){ return{ message: "Hello World!" }; } }; Vue.createApp(app).mount("#app"); </script> </body> </html>