RNA-seqその8、edgeRを使ったTMM正規化
21 June 2019追記
文章を少し修正した
追記ここまで
featrureCountの結果をedgeRを使って正規化する
このtutorialのpdfファイルと以下のリンク先を参考にした
Statistical analysis of RNA-Seq data
http://www.nathalievialaneix.eu/doc/pdf/tutorial-rnaseq.pdf
ここにPDFのリンクがある
NV² | Biostatistics analysis of RNA-Seq data
実際の処理方法はこちらの方法を
参考にさせてもらった
edgeR - macでインフォマティクス
シロイヌナズナのRNA seq解析 - macでインフォマティクス
edgeRのユーザーズガイドも必読
edgeR: リードカウントから発現変動遺伝子を検出 - Heavy Watal
以前書いた正規化についてのメモ
mecobalamin.hatenablog.com
これまでにやったのは以下の処理
- シークエンスデータのダウンロード
- trimmomaticでデータのクリーニング
- Hisat2でマッピング
- featureCountでリードをカウント
NCBIから2種類の細胞種について
3つずつファイルをダウンロードして
それぞれに上記と同様の処理をした
本来ならbiological replicate間で比較をするそうだが、
処理に使ったデータは細胞種が同じなだけで
実験的には無関係のデータであり、
"biological replicateではない"
よくないとは思うけど、スクリプトの確認のため
これらを使って正規化までの処理を確認する
なお6個ファイル処理するのに
手持ちのPCで数日かかった
1TBの外付けHHDでは全く足りず
中間ファイルを消しつつ処理した
まずRを使ってカウントしたファイルの中身を確認する
すでに読み込まれているコマンドや関数を削除して
edgeRのライブラリの読み込む
グラフはggplotを使わずに表示している
見た目の問題なので表示は参照先と同じにできているはず
rm(list = ls(all = T)) library(edgeR)
つぎにリードカウントのファイルを読み込む
# 表示サイズを設定 options(digits = 4, width = 120) # counts.txtのあるディレクトリのパス setwd("c:\\path\\to\\countfile") # リードカウントのファイルを読み込む Results.FC <- read.table("counts.txt", header = TRUE, skip = 1, sep="\t") Names.ID <- c("Geneid", "Chr", "Start", "End", "Strand", "Length", "H1", "H2", "H3", "J1", "J2", "J3") colnames(Results.FC) <- Names.ID rownames(Results.FC) <- sub("\\.[0-9]{1,}$", "", as.character(Results.FC[, 1])) Count.Data <- as.matrix(Results.FC[, 7:12])
H1, H2, H3及びJ1, J2, J3はカウントの列につけた任意の名前
HとJで細胞種が異なる
featureCountの出力では読み込んだbamファイルの名前がついているが
わかりやすくするために書き換える
行名はGeneidからピリオド以下を削除して使う
カウントデータは7-12列目でCount.Dataを計算に使う
Results.FCの中身を確認するとこんな感じ
> head(Results.FC) Geneid Chr NR_046018 NR_046018.2 chr1;chr1;chr1 NR_024540 NR_024540.1 chr1;chr1;chr1;chr1;chr1;chr1;chr1;chr1;chr1;chr1;chr1 NR_106918 NR_106918.1 chr1 XR_001737835 XR_001737835.1 chr1;chr1;chr1 NR_036051 NR_036051.1 chr1 NR_026818 NR_026818.1 chr1;chr1;chr1 Start NR_046018 11874;12613;13221 NR_024540 14362;14970;15796;16607;16858;17233;17606;17915;18268;24738;29321 NR_106918 17369 XR_001737835 29926;30564;30976 NR_036051 30366 NR_026818 34611;35277;35721 End NR_046018 12227;12721;14409 NR_024540 14829;15038;15947;16765;17055;17368;17742;18061;18366;24891;29370 NR_106918 17436 XR_001737835 30039;30667;31295 NR_036051 30503 NR_026818 35174;35481;36081 Strand Length H1 H2 H3 J1 J2 J3 NR_046018 +;+;+ 1652 0 0 0 0 0 8 NR_024540 -;-;-;-;-;-;-;-;-;-;- 1769 10 35 199 0 13 1198 NR_106918 - 68 0 0 6 0 0 149 XR_001737835 +;+;+ 538 0 0 0 0 0 0 NR_036051 + 138 0 0 0 0 0 1 NR_026818 -;-;- 1130 0 0 0 0 0 0
正規化をする関数は以下の通り
Statistical analysis of RNA-Seq dataのRaw data filteringのところで
カウントの合計が0以上の行を残しているのでこれを使った
参考にしたサイトでは違うようだ
リードカウントの下限の決め方は色々ありそうなので今後試す
TMMNormalization <- function(Count.Data){ #Filtering #リードカウントが0以上を残す Count.Keep <- rowSums(Count.Data) > 0 Count.Data <- Count.Data[Count.Keep, ] Count.Data.log <- log2(Count.Data + 1) WriteGraphs(Count.Data, Count.Data.log, "Original") Names.Group <- factor(c("positive", "positive", "positive", "negative", "negative", "negative")) # edgeRを使った正規化係数の計算 Count.DGE <- DGEList(counts = Count.Data, group = Names.Group) Count.DGE <- calcNormFactors(Count.DGE) Count.DGE <- estimateCommonDisp(Count.DGE) Count.DGE <- estimateTagwiseDisp(Count.DGE) Results.DGE <- exactTest(Count.DGE) Table.DGE <- as.data.frame(topTags(Results.DGE, n = nrow(Count.Data))) Tag.DGE <- as.logical(Table.DGE$FDR < 0.05) Names.DGE <- rownames(Table.DGE)[Tag.DGE] # 正規化係数とライブラリサイズの取得 Norm.Factors <- Count.DGE$samples$norm.factors Lib.Size <- Count.DGE$samples$lib.size # 正規化と対数の計算 Count.Norm <- t(apply(Count.Data, 1, function(x) x / (Norm.Factors * Lib.Size) * 10^6)) Count.Norm.log <- log2(Count.Norm + 1) WriteGraphs(Count.Norm, Count.Norm.log, "TMMNorm") return(Count.Norm.log) }
正規化前のカウント
>print(head(Count.Data.log)) H1 H2 H3 J1 J2 J3 NR_046018 0.000 0.00 0.000 0 0.000 3.170 NR_024540 3.459 5.17 7.644 0 3.807 10.228 NR_106918 0.000 0.00 2.807 0 0.000 7.229 NR_036051 0.000 0.00 0.000 0 0.000 1.000 XR_001737584 0.000 0.00 0.000 0 0.000 3.585 XR_001737581 0.000 0.00 0.000 0 0.000 3.585
カウントの合計が0の行が除かれている
で、正規化されるとこうなる
>print(head(Count.Norm.log)) H1 H2 H3 J1 J2 J3 NR_046018 0.0000 0.000 0.00000 0 0.0000 0.011020 NR_024540 0.1552 1.109 0.94713 0 0.5704 1.103176 NR_106918 0.0000 0.000 0.03981 0 0.0000 0.192592 NR_036051 0.0000 0.000 0.00000 0 0.0000 0.001382 XR_001737584 0.0000 0.000 0.00000 0 0.0000 0.015131 XR_001737581 0.0000 0.000 0.00000 0 0.0000 0.015131
DGEListでedgeRで使う形式にデータを変えている
正規化係数やライブラリのサイズも出力されている
Count.DataのH1のカウントについてヒストグラム出力

正規化前の分布をbox plotで表示する
元のデータがバラバラだからしょうがないか
ここはまだ理解が怪しいところなのだが、
TMM正規化にはMA plotで閾値を決めて
残ったリードカウントを使っているっぽい
H1とH2のリードカウントをMA plot表示するとなんとなくそれっぽい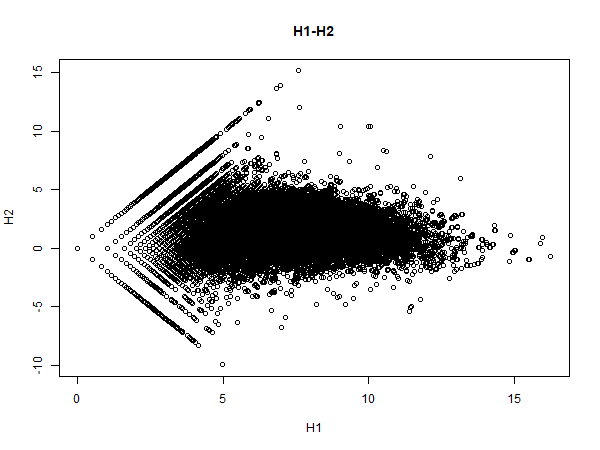
外れ値?
除く必要がありそうだけどやり方がわからないので
とりあえずそのまま処理を続ける
全部組み合わせる必要はなさそうだけどコードの関係。
MAPlot <- function(d){ c <- colnames(d) for(i in c){ for(j in c){ if(i != j){ M = d[,i] - d[,j] A = (d[,i] + d[,j]) / 2 df <- data.frame(A, M) print(head(df)) png(sprintf("MAplot_%s-%s.png", i, j), width = 600, height = 450) plot(df, xlab = i, ylab = j, main = sprintf("%s-%s", i, j)) dev.off() } } } }
edgeRの関数を使ってカウント全部をプロットすると
なんか違う形の分布になった
plotSmear(Results.DGE, de.tags. = Names.DGE, cex = 0.3)
リードカウントをライブラリサイズと正規化係数で正規化し、box plot表示する
外れ値が増えたようだがどうなんだろう
ヒストグラムとbox plotの出力には以下の関数を使っている
WriteGraphs <- function(d.norm, d.log, g.tag){ h.tag <- "H1" # グラフの保存 # ヒストグラムの表示 png(sprintf("histogram_count_norm_%s_%s.png", g.tag, h.tag), width = 600, height = 450) hist(d.norm[, h.tag], xlab = h.tag, ylab = "count", main = "norm") dev.off() #対数 png(sprintf("histogram_count_log_%s_%s.png", g.tag, h.tag), width = 600, height = 450) hist(d.log[, h.tag], xlab = h.tag, ylab = expression(log[2](count + 1)), main = "log") dev.off() #box plot #対数 png(sprintf("boxplot_count_log_%s.png", g.tag), width = 600, height = 450) boxplot.matrix(d.log, ylab = expression(log[2](count + 1))) dev.off() }
試しにEGFRの数を比較する
正規化したデータから目的のAccession Numberを探して
カウント数を調べる
FindGeneInterest <- function(d){ #find genes of interest acc.egfr <- c("NM_005228", "NM_201282", "NM_201283", "NM_201284", "NM_001346897", "NM_001346898", "NM_001346899", "NM_001346900", "NM_001346941") iso.egfr <- c("a", "b", "c", "d", "e", "f", "g", "h", "i") var.egfr <- t(rbind(acc.egfr, iso.egfr)) egfr <- c() for(i in var.egfr[,1]){ egfr <- rbind(egfr, d[i,]) } print(data.frame(var.egfr, egfr)) }
これが出力
acc.egfr iso.egfr H1 H2 H3 J1 J2 J3 1 NM_005228 a 2.707 5.064 5.414 0 0 0 2 NM_201282 b 1.247 3.233 4.108 0 0 0 3 NM_201283 c 0.710 2.865 3.213 0 0 0 4 NM_201284 d 1.261 3.238 4.120 0 0 0 5 NM_001346897 e 1.811 4.116 5.002 0 0 0 6 NM_001346898 f 1.830 4.116 5.006 0 0 0 7 NM_001346899 g 2.697 5.064 5.412 0 0 0 8 NM_001346900 h 2.684 5.038 5.403 0 0 0 9 NM_001346941 i 2.638 5.012 5.252 0 0 0
前後するがこっちが正規化前の対数でないリードカウント
acc.egfr iso.egfr H1 H2 H3 J1 J2 J3 1 NM_005228 a 487 981 8931 0 0 0 2 NM_201282 b 121 254 3482 0 0 0 3 NM_201283 c 56 190 1774 0 0 0 4 NM_201284 d 123 255 3514 0 0 0 5 NM_001346897 e 221 494 6658 0 0 0 6 NM_001346898 f 225 494 6675 0 0 0 7 NM_001346899 g 483 981 8914 0 0 0 8 NM_001346900 h 478 963 8861 0 0 0 9 NM_001346941 i 460 945 7957 0 0 0
HとJはともに培養細胞のシークエンス結果で、
HにはEGFRの発現があり、Jには発現していないと言われている
実際その通りになっているように見える
H1とH3では桁が変わるほどカウント数に違いがあったが
正規化により桁はだいたい揃ってる
似たような発現量ってことでいいのだろうか
もっともMA plotではシークエンスの結果が不均一っぽい
正規化が正しく計算できていないと思う
なのでこのまま数字の比較はできないかも
そもそもreplicateでないシークエンスのデータを
無理やりまとめて正規化してるからからか
うまく外れているところを除くか使うデータを変えてみるか。。。
処理の流れとしてはこれで良さそうではあるが。。。
コードをまとめる
rm(list = ls(all = T)) library(edgeR) WriteGraphs <- function(d.norm, d.log, g.tag){ h.tag <- "H1" # グラフの保存 # ヒストグラムの表示 png(sprintf("histogram_count_norm_%s_%s.png", g.tag, h.tag), width = 600, height = 450) hist(d.norm[, h.tag], xlab = h.tag, ylab = "count", main = "norm") dev.off() #対数 png(sprintf("histogram_count_log_%s_%s.png", g.tag, h.tag), width = 600, height = 450) hist(d.log[, h.tag], xlab = h.tag, ylab = expression(log[2](count + 1)), main = "log") dev.off() #box plot #対数 png(sprintf("boxplot_count_log_%s.png", g.tag), width = 600, height = 450) boxplot.matrix(d.log, ylab = expression(log[2](count + 1))) dev.off() } MAPlot <- function(d){ c <- colnames(d) for(i in c){ for(j in c){ if(i != j){ M = d[,i] - d[,j] A = (d[,i] + d[,j]) / 2 df <- data.frame(A, M) print(head(df)) png(sprintf("MAplot_%s-%s.png", i, j), width = 600, height = 450) plot(df, xlab = i, ylab = j, main = sprintf("%s-%s", i, j)) dev.off() } } } } FindGeneInterest <- function(d){ # find genes of interest acc.egfr <- c("NM_005228", "NM_201282", "NM_201283", "NM_201284", "NM_001346897", "NM_001346898", "NM_001346899", "NM_001346900", "NM_001346941") iso.egfr <- c("a", "b", "c", "d", "e", "f", "g", "h", "i") var.egfr <- t(rbind(acc.egfr, iso.egfr)) egfr <- c() for(i in var.egfr[,1]){ egfr <- rbind(egfr, d[i,]) } print(data.frame(var.egfr, egfr)) } TMMNormalization <- function(Count.Data){ # Filtering # リードカウントが0以上を残す Count.Keep <- rowSums(Count.Data) > 0 Count.Data <- Count.Data[Count.Keep, ] Count.Data.log <- log2(Count.Data + 1) #print(head(Count.Data.log)) # グラフの出力 MAPlot(Count.Data.log) WriteGraphs(Count.Data, Count.Data.log, "Original") Names.Group <- factor(c("positive", "positive", "positive", "negative", "negative", "negative")) # edgeRを使った正規化係数の計算 Count.DGE <- DGEList(counts = Count.Data, group = Names.Group) Count.DGE <- calcNormFactors(Count.DGE) Count.DGE <- estimateCommonDisp(Count.DGE) Count.DGE <- estimateTagwiseDisp(Count.DGE) Results.DGE <- exactTest(Count.DGE) Table.DGE <- as.data.frame(topTags(Results.DGE, n = nrow(Count.Data))) Tag.DGE <- as.logical(Table.DGE$FDR < 0.05) Names.DGE <- rownames(Table.DGE)[Tag.DGE] # plotSemerを使ってMA plotの出力 png("MAplot_plotSemer_DGEresults.png", width = 600, height = 450) plotSmear(Results.DGE, de.tags. = Names.DGE, cex = 0.3) dev.off() # 正規化係数とライブラリサイズの取得 Norm.Factors <- Count.DGE$samples$norm.factors Lib.Size <- Count.DGE$samples$lib.size # 正規化と対数の計算 Count.Norm <- t(apply(Count.Data, 1, function(x) x / (Norm.Factors * Lib.Size) * 10^6)) Count.Norm.log <- log2(Count.Norm + 1) #print(head(Count.Norm.log)) # グラフの出力 WriteGraphs(Count.Norm, Count.Norm.log, "TMMNorm") return(Count.Norm.log) } options(digits = 4, width = 120) setwd("c:\\path\\to\\Count") Results.FC <- read.table("counts.txt", header = TRUE, skip = 1, sep="\t") Names.ID <- c("Geneid", "Chr", "Start", "End", "Strand", "Length", "H1", "H2", "H3", "J1", "J2", "J3") colnames(Results.FC) <- Names.ID rownames(Results.FC) <- sub("\\.[0-9]{1,}$", "", as.character(Results.FC[, 1])) Count.Data <- as.matrix(Results.FC[, 7:12]) Data.Norm <- TMMNormalization(Count.Data) FindGeneInterest(Data.Norm)
一部Atomでスクリプトを実行するために
必要な書き方になっている
ここのprint()など
print(head(d))
はRGuiではprintは必要ない