RNA-seqその3、trimmomatic
分割したFastqファイルのクリーニングをする
mecobalamin.hatenablog.com
trimmomaticについて
Trimmomatic | FASTQ クリーニングツール
trimmomaticを実行するとファイルへのアクセスがとても多い
windowsのタスクマネージャーを見ると
解析するデータが大きいのもあって
ディスクが100%近くでアクティブになる
HDDには良くない感じに見える
まず下準備としてRAMディスクを用意する
これは壊れても交換しやすいディスクがあれば別にいらないと思う
ubuntuには標準でRAMディスクがあるが、
wslのtmpfsはエミュレーションしているだけらしい
Does tmpfs really work? : bashonubuntuonwindows
なのでwindowsで動くRAMディスクをインストールする
「ImDisk Virtual Disk Driver」システムメモリ上に仮想ディスクを作成できるツール - 窓の杜
インストールしたけど場所がわからない。。。。
コントロールパネルで表示を小さいアイコンにすると
ImDisk Virtual Disk Driverのアイコンが見つかるので
ショートカットをデスクトップに作っておく
PCを再起動後にImDiskを実行する
wslを起動した状態だとマウントできないみたい
10GのRAMディスクを作成
ドライブ名はR


wslのターミナルを起動して
dfコマンドでRAMディスクにマウントできていることを確認する
マウント先はImDiskで設定したR
$ df Filesystem 1K-blocks Used Available Use% Mounted on R: 10485756 1432936 9052820 14% /mnt/r
ImDiskでボリュームを削除してもマウントが残ってたりする
RAMディスクを作ったり消したりしたらPCの再起動をしとくのが良さげ
RAMディスクを作ってマウントできてなければ手動でマウントする
それとImDiskで削除する前にアンマウントする
それぞれのコマンドはこれ
$ sudo mount -t drvfs r: /mnt/r $ sudo umount /mnt/r
作ったRAMディスクをテンポラリディスクとして使用する
ここから本題
trimmomaticの処理は以下の流れで行う
- copy files to tmp directory
- fastqc
- trimmomatic
- fastq-stats
- remove unnecessary string
- prinseq-lite
trimmomaticの前後でやっているのはシークエンスデータの統計データの取得
クリーニングの前後でデータの比較を行うための処理をやっている、
そうなんだけど見てもまだよくわからない。。。。
とりあえず使ったスクリプトだけ掲載
アダプターが取り除かれた配列が
"trim_"のついたディレクトリに保存される
#!/bin/bash cd $HOME/usr/scripts/ w_dir="$HOME/usr/data" scripts_dir="$HOME/usr/scripts" tmp_dir="/mnt/r/tmp" list_name="list_sra.txt" list_dir="${scripts_dir}" list_raw=$(cat ${list_dir}/${list_name} | wc -l) list_file=$(cat ${list_dir}/${list_name} | sed 's/\.sra//g' ) postfix="fastq" quality="15" adapter="$HOME/miniconda3/share/trimmomatic-0.38-1/adapters/TruSeq3-PE.fa" for i in ${list_file} do echo "analyze ${i}" trim_dir="${w_dir}/trim_${i}/trim" stats_dir="${w_dir}/trim_${i}/stats" fastqc_dir="${w_dir}/trim_${i}/fastqc" prinseq_dir="${w_dir}/trim_${i}/ps" mkdir -p ${trim_dir} mkdir -p ${stats_dir} mkdir -p ${fastqc_dir} mkdir -p ${prinseq_dir} mkdir -p ${tmp_dir} in_dir="${w_dir}/raw/div_${i}" div_list="${in_dir}/list_div_${i}.txt" infile_1=$(grep _1_ ${div_list}) for j in ${infile_1} do infile_2=${j/_1_/_2_} logfile=${j/_1_/_} echo "copy files to tmp directory" cp ${in_dir}/${j} ${tmp_dir}/ cp ${in_dir}/${infile_2} ${tmp_dir}/ echo "fastqc began" for k in ${j} ${infile_2} do fastqc --nogroup -o ${fastqc_dir} ${tmp_dir}/${k} done echo "finished." echo "trimmomatic began" trimmomatic \ PE \ ${tmp_dir}/${j} \ ${tmp_dir}/${infile_2} \ ${tmp_dir}/trim${quality}_${j}.${postfix} \ ${tmp_dir}/trim${quality}_${j}.${postfix}-unique.out \ ${tmp_dir}/trim${quality}_${infile_2}.${postfix} \ ${tmp_dir}/trim${quality}_${infile_2}.${postfix}-unique.out \ ILLUMINACLIP:${adapter}:2:30:10 \ LEADING:3 \ TRAILING:3 \ SLIDINGWINDOW:4:${quality} MINLEN:36 \ &> ${trim_dir}/trim_${logfile}.log echo "finished." echo "fastq-stats began" for k in ${j} ${infile_2} do fastq-stats \ ${tmp_dir}/${k} >\ ${stats_dir}/raw_${k}.stats fastq-stats \ ${tmp_dir}/trim${quality}_${k}.${postfix} >\ ${stats_dir}/trim_${k}.stats done echo "finished." rm ${tmp_dir}/${j} rm ${tmp_dir}/${infile_2} echo "remove unnecessary string" for k in ${j} ${infile_2} do #sed -i".bak" '/length/s/^\+.*/+/' ${tmp_dir}/trim${quality}_${k}.${postfix} #mv ${tmp_dir}/trim${quality}_${k}.${postfix} ${tmp_dir}/rename_${k}.${postfix} #mv ${tmp_dir}/trim${quality}_${k}.${postfix}.bak ${tmp_dir}/trim${quality}_${k}.${postfix} cat ${tmp_dir}/trim${quality}_${k}.${postfix} | sed '/length/s/^\+.*/+/' \ > ${tmp_dir}/rename_${k}.${postfix} done echo "finished." mv ${tmp_dir}/trim${quality}_* ${trim_dir}/ echo "prinseq-lite began" prinseq-lite.pl \ -fastq ${tmp_dir}/rename_${j}.${postfix} \ -fastq2 ${tmp_dir}/rename_${infile_2}.${postfix} \ -out_format 3 \ -derep 24 \ -log ${prinseq_dir}/${logfile}.log \ -out_good ${tmp_dir}/ps_${logfile}_gd \ -out_bad ${tmp_dir}/ps_${logfile}_bd \ -trim_tail_right 5 \ -min_len 30 mv ${tmp_dir}/rename_* ${trim_dir} mv ${tmp_dir}/ps_${logfile}_* ${prinseq_dir} done rm -r ${tmp_dir} done
27 Feb. 2019修正
adapterのファイル名をSEからPEに修正した
14 April 2019追記
ファイルの容量がRAMディスクよりも大きいと
エラーを吐かずにスクリプトが止まる
今回は10GBでは足りず、16GBまで増やす必要があった
RAMディスクの容量を変えたあとに
PCの再起動をせずにスクリプトを実行したところ
ディスクをうまく認識できなかったみたいで
一瞬ファイルの書き込みがあって
実行中のまま反応がなくなった
その後が変な状況になってしまった
crtl + cでスクリプトは止められず
tmuxを終了してもjobが残っていたようだ
ターミナルを終了してPCの再起動をしても
ubuntuは生きていたようで強制終了するかの選択肢がでてきた
強制終了を選んでも再起動のまましばらく先に進まず。。。
結局電源ボタン長押しで再起動
一応PCは再起動してほかも問題なく動いてくれているけれど
次回もそうだとは限らないので
RAMディスク周りをいじるときは
その前後でPCとターミナルの再起動をしておく
そもそもRAMディスクを使うのは
データを入れているHDDの保護のためなので、
テンポラリに使えるディスクがあればいい
USBメモリとか?
気になるのは書き込み速度
あとで試す
追記ここまで
30 May 2019追記
余分な文字列を削除するコマンドのところで
やり直してみたらエラーが出る
sedのテンポラリファイルを移動か削除のときにパーミッションエラーが出てる
コメントアウトして別の方法でやってみたらうまくできたみたいなのでそっちも残す
追記ここまで
JBrowseでbamファイルを表示する
インストールしたJBrowseにゲノムデータを表示させる
表示させるのは
レファレンスにするヒトのゲノムデータと
GRCh38_latest_genomic.fna
ref_GRCh38.p12_top_level.gff3
HeLaのRNAseqのデータから作ったbamファイル
こっちが実験データの代わりになる
sort_SRR6799791.bam
sort_SRR6799791.bam.bai
の四種類
bamファイルの作り方は後ほど記録する
JBrowseへの登録はこっちに説明があるので
ここからやったことを記録してく
JBrowse Configuration Guide - GMOD
JBrowse FAQ - GMOD
まずはレファレンスデータの登録
jbrowseのprepare-refseqs.plを使う
bin/prepare-refseqs.pl --fasta <fasta file> [options]
実際に入力したのはこれ
$ cd /var/www/html/jbrowse $ bin/prepare-refseqs.pl --fasta ~/usr/data/db/GRCh38_latest_genomic.fna --out HeLa_SRR6799791_dev --trackLabel GRCh38_latest_genomic --seqType dna
"--out"オプションで表示する実験データにレファレンスを出力する
"--trackLabel"はJBrowse上での表示ラベル
"--seqType"はよくわからないけどゲノムデータなのでDNAにした
outオプションで指定したディレクトリに
seqというディレクトリが作られてその内部にも
大量のディレクトリとファイルが作られてく
結構時間が掛かる
wslを使っている場合はwindows defenderからのアクセスを切っておくほうが良い
ほっとくと書き込むファイル全部をスキャンしようとする
mecobalamin.hatenablog.com
次にgff3ファイルの登録
flatfile-to-json.plを使う
bin/flatfile-to-json.pl --[gff|gbk|bed] <flat file> --tracklabel <track name> [options]
入力したコマンドはこちら
$ bin/flatfile-to-json.pl --gff ~/usr/data/db/ref_GRCh38.p12_top_level.gff3 --trackType CanvasFeatures --out HeLa_SRR6799791_dev --trackLabel ref_GRCh38
bamとbam.baiの登録もスクリプトを使う
スクリプト名はadd-bam-track.pl
baiファイルはbamと同じディレクトリに入れておけば
自動で認識してくれる
bamとbam.baiはbamという名前のディレクトリに入れて
HeLa_SRR6799791_dev以下に保存する
add-bam-track.plはHeLa_SRR6799791_devの場所で実行する
$ cd HeLa_SRR6799791_dev $ ../bin/add-bam-track.pl --label HeLa_SRR6799791 --bam_url bam/sort_SRR6799791.bam --in trackList.json
これでとりあえず作業は終了
前回volvoxのデータを表示したようにwslでapache2を起動する
mecobalamin.hatenablog.com
windows側のブラウザから以下のアドレスにアクセスする
http://localhost/jbrowse/index.html?data=HeLa_SRR6799791_dev
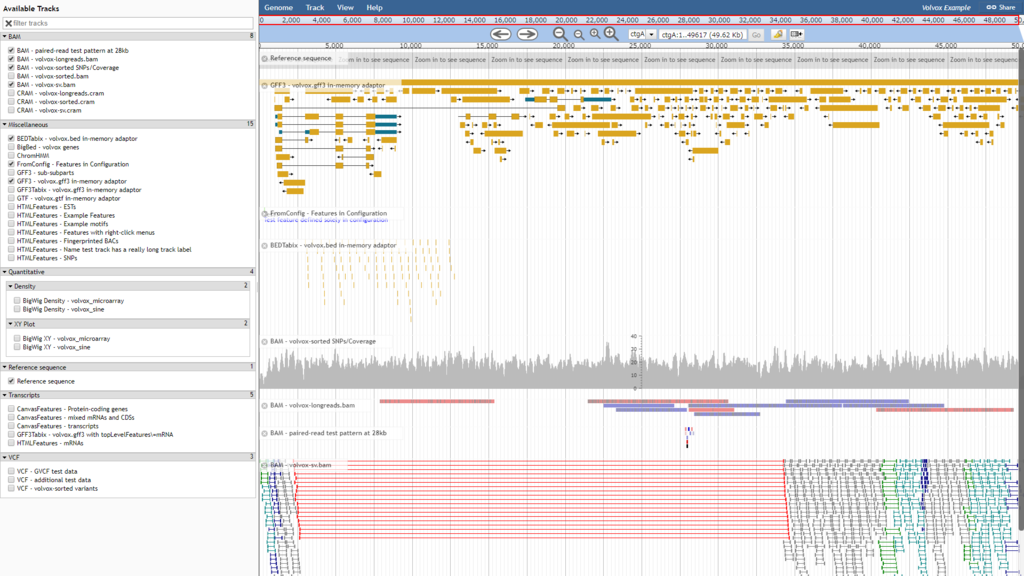
こんな感じで表示される
上段の黄色っぽく表示されているのがヒトゲノムデータ
下段の赤・青で表示されているのがRNAseqの結果

あまり一致していないようにみえる。。。
ヒトゲノムのコード領域と培養細胞で実際に転写されている領域は結構違うものなのか。。。?
そのあたり良くわからないけど、まぁ、とりあえず表示はできた
JBrowseのインストール、三度目の正直
12 October 2020 追記
mecobalamin.hatenablog.com
JBrowseをdockerで動くようにした
最低限必要なコマンドやライブラリをインストールした
以下の記事を参照する
mecobalamin.hatenablog.com
dockerが使えていればubuntuのコンテナに
JBrowseをインストールするのが
楽かもしれないと個人的には思った
追記ここまで
JBrowseのインストールをやりなおした。
変更点はインストール先を変えたこと、
サーバーをpythonのhttp.serverから
wslのapache2に変更したことの2点。
というのも作ったbamファイルが表示できなくて
いろいろ調べていたら公式の説明にこんなのがあった
JBrowse FAQ - GMOD
Note: servers like "SimpleHTTPServer" from Python or "http-server" from NPM are generally not full featured enough to run all JBrowse features correctly (SimpleHTTPServer does not support Range queries, and http-server interprets tabix files incorrectly). RangeHTTPServer does pass the test suite for jbrowse though, so it should work for tests https://github.com/danvk/RangeHTTPServer (but not for compressed json files).
pythonのSimpleHTTPServerではだめらしい
使っていたのはhttp.server。
サンプルデータのvolvoxは表示できたけど、
自分で作ったデータはうまく表示できないのもあったので
原因はよくわからないけどよくないのかも。。。
ということで
まずはapach2のインストールから。
Window10でLAMP (Fall Creators Update版) - Qiita
$ sudo apt install apache2
ちなみにLAMPとは、Linux、Apache、MySQL、PHP/Perl/Pythonのことだそう。
LAMP(Linux+Apache+MySQL+PHP/Perl/Python)とは - IT用語辞典 e-Words
LAMP (ソフトウェアバンドル) - Wikipedia
インストールしたapache2の起動と停止は次のコマンドで行う
$ sudo service apache2 start # 起動 $ sudo service apache2 stop # 停止
こういうやり方もあるようだけどまだ試してない
Apacheの起動 - Qiita
apache2を起動後windowsにインストールされた
web browserのアドレスバーに"localhost"と入れてエンターを押す。
apache2のindex.htmlが表示されればインストールがうまくいってる
/var/www/html/index.htmlが表示されている
次にnode.jsのインストール。
ダウンロードはここから
ダウンロード | Node.js
バイナリーファイルをダウンロードして展開
$ xz -d node-v10.16.0-linux-x64.tar.xz $ tar -xvf node-v10.16.0-linux-x64.tar
"/node-v10.15.0-linux-x64/bin/"に含まれる
node、npm、npxをパスの通っているディレクトリにリンクを貼る
コマンドは$HOME/binにリンクを貼るようにしてて
.bashrcにパスを書いてある
$HOME/binにリンクを貼るときはlnコマンドを使う
$ ln -s $HOME/local/node-v10.15.0-linux-x64/bin/node $HOME/bin
同様に"node-v10.15.0-linux-x64/lib/node_modules/yarn/bin/yarn"も
$HOME/binにリンクを貼る
あと、perlは/usr/bin/perlを使う。
miniconda3にもperlがあるが、
これを使ったら何故かコンパイルエラーが出ることがあった
どうもperlの設定ファイルのパス指定がおかしい様子。
なので.bashrcでminicondaよりも前に
/usr/bin/perlが来るようにパスを書く
export PATH="$HOME/bin:/usr/bin:$HOME/miniconda3/bin:$PATH"
これらの準備が済んでからJBrowseのインストールをする
前回よりもバージョンがちょっとだけ上がってる
任意のディレクトリにアーカイブをダウンロードして展開する
今回はホームディレクトリのtmpにwgetを使ってダウンロードする
unzipで展開後してできたディレクトリを
/var/www/html/に移動する
$ cd ~/tmp $ wget https://github.com/GMOD/jbrowse/archive/1.16.3-release.zip $ unzip 1.16.3-release.zip $ sudo mv jbrowse-1.16.3-release/ /var/www/html/jbrowse
もしjbrowse/のオーナーがrootのままなら所有者を変える
インストールでsetup.shを実行するが、sudoで実行してはいけないらしい
ちなみにwhoamiは自分のユーザー名に変える
$ sudo chown -R 'whoami' jbrowse
そしてjbrowse内のsetup.shを実行する
$ cd jbrowse
$ ./setup.sh
apacheを起動してwindowsのweb browseから
volvoxのデータにアクセスできたらインストール成功
http://localhost/jbrowse/index.html?data=sample_data%2Fjson%2Fvolvox
ここまで書いてなんだけど
IGVとか使えばlocal serverを立てなくても
表示できる
というかこっちが楽
サーバー立てたら共有できる点が便利
Home | Integrative Genomics Viewer
これで自作のファイルが表示できるようになればいいのだが。
25 Feb. 2019追記
apache2の起動時にエラーが表示されてた
(92)Protocol not available: AH00076: Failed to enable APR_TCP_DEFER_ACCEPT
ググると解決法があったので真似してファイルに追記した
【BoUoW】Apache起動時の「Failed to enable APR_TCP_DEFER_ACCEPT」 - Qiita
編集したファイルはapache2.conf
$ sudo vim /etc/apache2/apache2.conf
ファイルの最後に以下の二行を追加
AcceptFilter http none AcceptFilter https none
それとapache2で立てたサーバーに
ローカルネットワークからアクセスしたい
Apacheをローカルネットワークのみに公開にする | EasyRamble
サーバーになっているPCのIPアドレスと開放するポートを
ports.confに記入すればいいっぽい
$ sudo vim /etc/apache2/ports.conf
ports.confの最後の行に追記した
Listen 192.168.100.110:8080
windowsでIPアドレスを確認するには
[スタートを右クリック] -> [設定] ->
[ネットワークとインターネット] -> [Wi-Fi]
とクリックする
次に利用しているWi-FiまたはLANの名前をクリックすると
つながっているネットワークのプロパティが確認できる
IPv4アドレスをListenから:8080までの間に記入する
一応同一ネットワークにあるタブレットからvolvoxのサンプルデータを見れた
アドレスは"localhost"をファイルに追加したIPアドレスとポートに書き換える
http://192.168.100.110:8080/jbrowse/index.html?data=sample_data%2Fjson%2Fvolvox
追記ここまで
31 May 2019 追記
インストールでエラーが出る場合の対応について
ライブラリが足りなくてエラーが出る場合がある
公式のサイトに対応方法があるが、libgd2-xpm-devは見つからない
JBrowse Troubleshooting - GMOD
代わりにlibgd-devをインストールするとうまくいった
$ sudo apt-get install build-essential libpng-dev zlib1g-dev libgd-dev
追記ここまで
Atomでscript
普段使っているエディターはAtom
殆どいじっていないが、
pythonとRを実行できるようにしてある
必要なpackageは
scriptとlanguage-r
packageをインストールしたら
windowsのシステム環境変数で
Rscriptのディレクトリにパスを通す
\R-3.5.0\bin
windowsの設定 -> 環境変数を検索 -> システムのプロパティ -> 環境変数
または
コントロールパネル -> システム -> システムのプロパティ -> 環境変数
からpathを編集する
やってたはずだけど
windows10を再インストールしたときに
やり忘れてたみたいなので
これどっかに書いてあったはずだけど。。。
あとlanguage-rはそのままだとエラーが出たはず
ファイルの記入ミスだったと思う
修正方法を載せたサイト見つけたら追記予定
27 May 2019追記
まずcummulative maxでエラーが出る件についてはここを参考に修正する
Atom editor r-language error - Failed to load snippets - Stack Overflow
Duplicate key 'Cummulative max' · Issue #17 · REditorSupport/atom-language-r · GitHub
次にgrepのところでエラーが出るが、
同じキーが有るのが良くないっぽい
'Grep': 'prefix': 'grep' 'body': 'grep(${1:pattern}, ${2:x}, ${3:ignore.case = ${4:FALSE}}, ${5:perl = ${6:FALSE}})' 'Grep': 'prefix': 'grep' 'body': 'grep(${1:pattern}, ${2:x}, ${3:ignore.case = ${4:FALSE}}, ${5:perl = ${6:FALSE}}, ${7:value = ${8:FALSE}}, ${9:fixed = ${10:TRUE}})'
これでとりあえずエラーは出なくなった
追記ここまで
16 October 2020 追記
Atomの出力に日本語を表示する
言語ごとに設定が違うっぽいが
pythonの場合は
[file] -> [initial script] -> init.coffee]
に
process.env.PYTHONIOENCODING = "utf-8";
を追記する
追記ここまで
メモ: コマンドのリンク先とWSLのCPU使用率
condaでパッケージをインストールするときの注意点
pythonとパッケージをインストールしたはずなんだけど
コマンド実行中にモジュールが見つからないってエラーが出ることがある
実際Trinityを使っててnumpyがないってエラーがでた
パッケージを入れてあるか確認。
$ conda list
numpyの1.15.4がインストールされてる。
pythonは3.6.8
pythonを実行して確認するとバージョンが違う
$ python --version
Python 2.7.15rc1
/usr/binにインストールされてるpythonを使っていたようだ
ubuntuには予めpythonがインストールされてて
condaで入れたpython/packageとは別になる
さてどうしよう。。。
このときは~/bin以下にminiconda3で入れたpythonのリンクを貼った
~/.bashrcにこんな感じでpathを記入して~/binにpathを通す
すでに書かれている場合は追記で。
前ほど優先順位が高いので、他のプログラムで使っているコマンドとの兼ね合いもある
export PATH="$HOME/bin:/usr/bin:$HOME/miniconda3/bin:$PATH"
~/binにpythonリンクを貼る
$ ln -s $HOME/miniconda3/bin/python $HOME/bin
これでcondaで入れたpythonとパッケージを使えるようになる
それとWSLの問題について
WSLでコマンドを実行中にwindowsのタスクマネージャーを見ると
CPUの使用率がやたらと高い時がある
perlだったりubuntuだったりならわかるが
windows defender antivirus serviceの使用率が
30-40%程度になることがある
ググるとwindowsに以前からある問題らしい
ここを参考に設定をした
Windows10の重い遅い対策 - Qiita
やったのはMsMpEng.exeを検疫対象から外して
WSLのdataが入るディレクトリを丸ごと除外した
書き込んだファイルをすべてチェックしようとするため
書き込みの多いコマンドを使うとチェックが続いて
結果CPUの使用率が上がる
Trinityでは大量にfastqファイルが生成されるらしく
それがいちいち検疫される
この設定の結果、windows defender antivirus serviceの
CPU使用率がやや下がった
常に使用率1番だったのが順位が入れ替わるようになって
10-20%台まで落ちてる
少しでも他にリソースが回せるようになったのは助かる
RNA-seqその2、データのダウンロードと変換
シークエンスのデータの入手方法には
- 公共のデータベースからシークエンスデータをダウンロードする
- シークエンスを行う
などがある。
今回は公共のデータベースのデータを使う
公共データベースによって登録されているファイル形式は異なる
NCBIはSRA、DDBJ/EMBLEはfastq形式である
シークエンスの結果は一般にはfastq形式で
SRAはFastqに情報を追加して圧縮した形式である
解析にはFastq形式を使用するので、ツールを用いてSRAからFastqに変換をする必要がある。
解析に使用するシークエンスデータが複数ある場合
ファイル名を記入したリストファイルの作成してあると一括で処理できる。
まずはダウンロード。
Home - SRA - NCBI
例題としてHeLaのRNAseqを探す
検索窓に"HeLa RNAseq"と入れて検索すると312個引っかかった
試しにこのデータを使ってみる
RNA-seq of HeLa cells - SRA - NCBI
しかし、データの評価の仕方がまだよくわからない
これを選んだのはなんとなく。。。
データのアクセッションナンバーはSRR6799791で、
アドレスはここで探せる
SRA Explorer
実際に探したアドレスを使ってファイルをダウンロード
$ cd $HOME/usr/data/ $ mkdir ./sra/ $ cd ./sra/ $ wget ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR679/SRR6799791/SRR6799791.sra
サイズが2.9 GBあるので時間がそれなりに掛かる
"$HOME"は、チルダで書くのがいいんだろうけど
スクリプト内で"$HOME"を使っているのでこの表記で。
SRA Toolkitを入れてればコマンドも使えるようだけど
使ったことがないので割愛
SRA Toolkit 使い方 公開データのダウンロードとsra fastq変換 – バイオインフォ 道場 [bioinfo-Dojo]
ダウンロードしたファイルはSRA Toolkitsに含まれる
fastq-dumpを使ってfastqに変換する
SRA Toolkitは前回インストール済み
RNA-seqその1、インストール - mecobalamin’s diary
16 April 2020 追記
SRA Explorerで生成したアドレスではダウンロードできなくなっているっぽい
SRA toolkitのprefetchでダウンロードする
prefetchを実行する前にvdb-confingを実行する
Toolkit Documentation : Software : Sequence Read Archive : NCBI/NLM/NIH
vdb-configのあるディレクトリで以下のように実行する
./vdb-config -i
RAMのサイズ以外は変更していない
追記ここまで
fastq-dumpで変換しただけでは動かないコマンドがあったりする
実際Trinityが動かなかった
NCBI SRA Toolkitの使い方 - アメリエフのブログ
Trinityはアセンブルを行うツール
詳しくはまた今度。
で、sraファイルを複数置換できるようにスクリプトを書いた
まずsraファイルのリストを作成
$ ls $HOME/usr/data/sra/ > $HOME/usr/scripts/list_sra.txt
以下のスクリプトこの名前"RNAseq01_SratoFastq.sh"で保存
保存場所は$HOME/usr/scripts/
#!/bin/bash echo "change directory to $HOME/usr/scripts/" cd $HOME/usr/scripts/ w_dir="$HOME/usr/data" scripts_dir="$HOME/usr/scripts" sra_dir="${w_dir}/sra" out_dir="${w_dir}/raw" list_name="list_sra.txt" list_dir="$HOME/usr/scripts" echo "making directory" mkdir -p ${out_dir} echo "run fastq-dump" for i in $(cat ${list_dir}/${list_name} | sed 's/\.sra//g') do file=${list_dir}/${i}.sra fastq-dump \ --split-files \ --defline-seq '@$sn[_$rn]/$ri' \ --outdir ${out_dir} \ ${sra_dir}/${i}.sra sed -i -e 's/_forward//' ${out_dir}/${i}_1.fastq sed -i -e 's/_reverse//' ${out_dir}/${i}_2.fastq done
"--defline-seq"でRead1とRead2の区別がつくように、
配列名の最後に/1または/2をつけている
NCBI SRA Toolkitの使い方 - アメリエフのブログ
元データがペアエンドのデータなので"--split-files"を指定する
シェルスクリプトの実行方法はこちら
$ cd $HOME/usr/scripts/ $ bash RNAseq01_SratoFastq.sh
ファイルに実行権限を与えて実行しても良い
$ cd $HOME/usr/scripts/ $ chmod 755 RNAseq01_SratoFastq.sh $ ./RNAseq01_SratoFastq.sh
chmodの実行は最初の一回だけ行う
【 chmod 】コマンド――ファイル/ディレクトリのパーミッション(許可属性)を変更する:Linux基本コマンドTips(14) - @IT
結果はrawに保存される
ファイル名はSRR6799791_1.fastq及びSRR6799791_2.fastqとなる
1、2には対になるリードがそれぞれ保存されている
十分にメモリとストレージがあれば、このままでいいのだが、
使っている環境に対してファイルサイズが大きすぎるのでファイルを分割する
fastq形式は1つのリードを4行で表示するので
4行単位で分割する
ここを参考にしてスクリプトを書いた
大きいfastqファイルを分割マッピング(tophat, linux) - script of bioinformatics
fastqを分割するツールもあった
fastq / fastaの操作ツール seqkit - macでインフォマティクス
とりあえずファイルを10分割する
ファイル名は"RNAseq02_divideFastqFiles.sh"
このスクリプトを実行すると10個に分割されたfastqと
ファイルリストのファイル"list_div_SRR6799791"が作成される
#!/bin/bash echo "change directory to $HOME/usr/data/raw/" cd $HOME/usr/data/raw/ scripts_dir="$HOME/usr/scripts" w_dir="$HOME/usr/data" out_dir="${w_dir}/raw" list_name="list_sra.txt" list_dir="$HOME/usr/scripts" n=10 for i in $(cat ${scripts_dir}/list_sra.txt | sed 's/\.sra//g') do div_file="div_${i}" echo "count row number" read=$(grep "\+${i}" $HOME/usr/data/raw/${i}_1.fastq | wc -l) sp=$(expr \( \( ${read} \/ ${n} \) + 1 \) \* 4) echo "making directory" mkdir -p $HOME/usr/data/raw/${div_file}/ echo "read=${read}, sp=${sp}" echo "dividing fastq file" for j in {1..2} do split --numeric-suffixes=01 -l ${sp} $HOME/usr/data/raw/${i}_${j}.fastq ${div_file}_${j}_ mv ${div_file}_${j}_* ./${div_file} done ls ${out_dir}/${div_file}/ > ${out_dir}/${div_file}/list_${div_file}.txt done
RNA-seqその1、インストール
wsl上でRNA-seqの解析を行う
手順を教えてもらったのでそのメモ
メモするのは
- コマンドのインストール(このページ)
- RNA-seqその2、データのダウンロードと変換 - mecobalamin’s diary
- RNA-seqその3、trimmomatic - mecobalamin’s diary
- RNA-seqその4、Fastqファイルのマージ - mecobalamin’s diary
- RNA-seqその5、Hisat2でマッピング - mecobalamin’s diary
- RNA-seqその6、featureCountsでリードカウント - mecobalamin’s diary
- RNA-seqその7、TrinityでアセンブルしてBlastにかける - mecobalamin’s diary
- RNA-seqその8、edgeRを使ったTMM正規化 - mecobalamin’s diary
- RNA-seqその9、DESeq2を使った正規化 - mecobalamin’s diary
- RNA-seqその10、Dockerで環境構築 - mecobalamin’s diary
教えてもらったのは1-7まで。
8と9はウェブから情報を集めて自分でやってみた。
それと表示に必要なJBrowseのインストールなど
- JBrowseのインストール、三度目の正直 - mecobalamin’s diary
- JBrowseでbamファイルを表示する - mecobalamin’s diary
- bigwigの作成と表示 - mecobalamin’s diary
- JBrowseにplug-inを追加する - mecobalamin’s diary
- hg38のfastaとgtf/gff3 - mecobalamin’s diary
ファイルの整形だったりクオリティのチェックだったり
他にもやっているけど大まかにはこんなところ
目的は実行に使うコマンドが動くコードを記録すること
教えてもらった元のコードはクラスターコンピュータで動いていた
そのコードをノートPCのwslで動くように書き換えた
コードの検証のため計算結果とグラフを載せるが、
結果の検証は実力不足でできてない(特に自分でやってみた正規化のあたり)
コードが動く ≠ 計算結果が正しい、ということに注意したい
Dockerで環境を統一できたら
結果の検証に集中できると思う(けどやってない)
ここではwslでの実行に必要なコマンドのインストールと設定を記載する
このあたりのサイトも参考にした
RNA-Seq | 遺伝子発現量解析
使用しているpcのスペック
CPU: Intel Core i5-7200U CPU @ 2.5 GHz
Memory: 24.0 GB (8 GB + 16 GB)
Storage: 250 GB SSD + 500 GB HDD -> 960 GB SDD + 1 T SSD
OS: Windows 10 Home version 1803 build 17134.523
WSL: 18.04.1 LTS
メモリを増設してあるがいたって普通のノートPC、
と思っているけどそうでもないらしい
【山田祥平のRe:config.sys】普通のPCが普通に買えない - PC Watch
CPUはこのスペックでも1日とかで終わってくれる
メモリとストレージは多いほどいいけどノートPCだとちょっと厳しいか
SSDも安くなってきてるからいずれ交換できればいいが。
それとwslはファイルのやり取りがものすごく遅いらしいので
HDDよりはSDD、外付けよりも内蔵のストレージを使いたいが問題がある
いくつかのコマンドはファイルの読み書きがとても多く
タスクマネージャーを見てるとSSDの寿命に影響しそう
1日中ディスクにアクセスとかやってくれる
なのでRAMディスク、外付けストレージをうまく使う
あとwslを使うときの注意点がある
ファイルシステムの関係でwindows側からwslのファイルを編集してはいけないらしい
実際ファイルがおかしくなったことがあった
今後のアップデートで変わる可能性があるらしいけど、現状は不可っぽい
出力されたファイルや設定ファイルなどをwindowsのエディタ等から編集しない
編集する場合はファイルをコピーして行う
次にコマンド・プログラムのインストールについて。
wslでインストールするコマンドと
windowsにインストールするプログラムとがある
wslにインストールするのは
windowsにインストールするのは
- R
- Rのライブラリ
まずwslで使用するコマンドのインストールする
アーカイブのダウンロードして、解凍、リンクを貼る、
ということをコマンド毎にする
コマンドのダウンロードはwgetを使う
【 wget 】コマンド――URLを指定してファイルをダウンロードする:Linux基本コマンドTips(24) - @IT
ダウンローダーは他にもあるようですが。。。
解凍は圧縮形式に合わせる
【 tar 】コマンド――アーカイブファイルを作成する/展開する:Linux基本コマンドTips(40) - @IT
【 unzip 】コマンド――ZIPファイルからファイルを取り出す:Linux基本コマンドTips(35) - @IT
リンクは$HOME/usr/localに展開したコマンドファイルから
パスの通っている$HOME/binにシンボリックリンクを貼っている
細かいルールを知らないので自己流かもだけど。。。
ちなみにwslでパスは$HOME/.bashrcに記入する
export PATH="$HOME/bin:$HOME/miniconda3/bin:$PATH"
ファイルがなければ作る
bashでパスを書く設定ファイルの選び方にも
なんかルールがあるっぽいがとりあえず。
パスは先に書かれている方から優先順位が高い
実際にコマンドをインストールする
- sra toolkit
NCBIにアップロードされている
シークエンスのデータはsra形式になっている
これを展開するにはsra toolkitを使う
ダウンロード先と参考にしたサイトはこちら
https://www.ncbi.nlm.nih.gov/sra/docs/toolkitsoft/
NCBI SRA Toolkitの使い方 - アメリエフのブログ
$ wget https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-ubuntu64.tar.gz
$ tar -zxvf sratoolkit.current-ubuntu64.tar.gz
シンボリックリンクを貼る
$ cp sratoolkit.2.9.2-ubuntu64/ ~/usr/local/ $ cd ~/bin $ ln -fs $HOME/usr/local/sratoolkit.2.9.2-ubuntu64/bin/fastq-dump ./
- prinseq-lite, FastQC, featureCount
prinseq-liteとFastQCもインストールしてリンクを貼る
prinseq-lite
PRINSEQ - Browse /standalone at SourceForge.net
PRINSEQ @ SourceForge.net
[Download] -> [Standalone] -> [prinseq-lite-0.20.4.tar.gz]をダウンロードする
prinseq-lite.plのパーミッションを変更する
FastQC
Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data
[Download Now] -> [FastQC v0.11.8 (Win/Linux zip file)]をダウンロードする
展開したファイルのfastqcのパーミッションを変更する
$ chmod 755 fastqc
featureCountも同様にインストールする
mecobalamin.hatenablog.com
- miniconda
次にminicondaを使ってpythonコマンドをインストールする
trimmomaticとtrinityはbiocondaのパッケージのひとつで、
minicondaをまずインストールして必要なパッケージを追加する
anacondaとminicondaの比較
https://echomist.com/anaconda-vs-miniconda/
minicondaは3.x系をインストールする
RNA-seqで使うコマンドによっては2.x系が必要な場合もあるが、
その時は2.x系の環境をつくってインストールする
念の為minicondaのpython3から$HOME/binにリンクを張って3.x系を使うようにしておく
$ ln -s $HOME/miniconda/bin/python $HOME/bin
minicondaの配布元とインストール用シェルスクリプトのアドレス
Miniconda — Conda documentation
https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh
シェルスクリプトをダウンロードして実行する
$ wget https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh $ bash Miniconda3-latest-Linux-x86_64.sh
実行後はターミナルを再起動してパスを読み込ませる
.bashrcにパスが書かれている場合はを.bashrcを再読込する
.bashrcの再読込のコマンドはこれ
$ source $HOME/.bashrc
- anaconda package, Trinity、fastq-stats、numpy、hisat2及びstringtie
パッケージはここからインストール元を探す
https://anaconda.org/
パッケージ名で検索をかけてダウンロード方法を探す
例えばtrimmomaticならインストール元はここで
Trimmomatic :: Anaconda Cloud
インストール方法を見るとこう書かれてる
$ conda install -c bioconda trimmomatic
biocondaはchannel名で、trimmomaticがpackage名で、
channelが登録されているかを確認するには
$ conda config --get channel
登録されてなければ次のようにして登録する
$ conda config --add channel <channel名>
チャンネルによってインストールされる内容が違っていたりする
一緒にインストールされるパッケージのバージョンが変わったり
インストールされたりされなかったりもある
必要なパッケージが揃っているかの確認も必要
追加するのはTrinity、ea-utils、fastq-stats、numpy、hisat2及びstringtie
$ conda install -c bioconda trinity $ conda install -c bioconda ea-utils $ conda install -c bioconda numpy $ conda install -c bioconda hisat2 $ conda install -c bioconda stringtie
- salmon
trinityでsalmonを使うが、biocondaから配布されているのは
バージョンが古い(v0.8.1)のでオリジナルの配布元から
binaryをダウロードして使う(v.0.12.0)
Overview – Salmon: Fast, accurate and bias-aware transcript quantification from RNA-seq data
$ wget https://github.com/COMBINE-lab/salmon/releases/download/v0.12.0/salmon-0.12.0_linux_x86_64.tar.gz
miniconda3/binにリンクを貼る
$ cp salmon-0.12.0_linux_x86_64/ $HOME/usr/local/ $ ln -s $HOME/usr/local/salmon-0.12.0_linux_x86_64/bin/salmon $HOME/miniconda3/bin/
先にv0.8.1のリンクがbinに貼られている場合はlnコマンドの前にリンクを解除する
$ unlink $HOME/miniconda/bin/salmon
29 May 2019追記
hisat2とstringtieを上記のコマンドでインストールすると
pythonのバージョンを2.7にしていたようだ
確認不足だった
この2つのコマンドはバイナリーを使ったほうがいいかも
hisat2
https://ccb.jhu.edu/software/hisat2/index.shtml
バイナリーのリンク
http://ccb.jhu.edu/software/hisat2/dl/hisat2-2.1.0-Linux_x86_64.zip
stringtie
StringTie
バイナリーのリンク
http://ccb.jhu.edu/software/stringtie/dl/stringtie-1.3.6.Linux_x86_64.tar.gz
それぞれwgetでダウンロードしてファイルを展開、
$HOME/binにシンボリックリンクを貼る
condaでインストールして使う場合はpython3とpython2の環境を分ける
別に記事を書く
mecobalamin.hatenablog.com
追記ここまで
23 April 2020 追記
Hisat2とstringtieをインストールしたら
python3にインストールされた
もしかしたら切り替えが必要なくなったのかも
追記ここまで
これで全部、のはず。。。
足りないのは適宜インストールする
インストールしたはずのパッケージが見つからない時がある
condaは3.xでそっちにインストールしているのに
wslに元から入っているpythonが2.xだったりして
パスの関係でwslのpythonで実行されていると見つからないってなる
以前そんな事があって、そのときはパスの優先順位を変えた
mecobalamin.hatenablog.com
pipを使う方法もあるが、anacondaと併用してはいけないそうだ
注意することかも
condaとpip:混ぜるな危険 - onoz000’s blog
次にwindowsで動かすプログラム
- R
Windows版をインストールする
The Comprehensive R Archive Network
RGuiか、AtomでR scriptでRのスクリプトを実行できるようにする
- edgeR
Rに必要なライブラリをインストールする
bioconductorを利用してインストールする
mecobalamin.hatenablog.com
- Atom (必要なら)
エディタで、パッケージの追加でRコマンドをスクリプトを書きつつ実行できる
atom.io
学生の頃はEmacsだったけど宗旨替え
wslではvi
/path/to/R/R-3.x.x/Rscript.exeにwindowsの環境変数でパスを通す
Rのアップデートをするたびに通し直す
mecobalamin.hatenablog.com
- script
- language-r
scriptでRを実行するのに必要
ATOMをR言語に対応させる方法 - Qiita
パッケージの更新が遅れているようで
設定ファイルにエラーが残ってるがそのままでも問題ない
修正方法もあったので直してあるけどそのサイトが見つからない。。。